
Диана Кондинская, руководитель отдела структурной биоинформатики BIOCAD, рассказывает про алгоритм AlphaFold2 от компании DeepMind.
Введение
Если вы когда-то слышали про биоинформатику, вычислительную биологию, структуру белка и проблему фолдинга, то вы знаете, о чем мы здесь будем рассказывать.
Если вы интересуетесь глубоким обучением и нейросетями, их применением для решения насущных практических задач и глубоко убеждены, что искусственный интеллект если и не завладеет миром, то точно превзойдет человека в своих когнитивных способностях, то об этой его победе над человеческим познанием вы тоже точно слышали.
Речь в нашей статье пойдет, как следует из названия, об искусственном интеллекте AlphaFold и его продолжателе AlphaFold2 от компании DeepMind, который был создан для предсказания трехмерной структуры белка. В 2020 году AlphaFold2 выполнил свою задачу так хорошо, как не удавалось никому до него, и с разгромными показателями победил своих соперников в соревновании, посвященном этой проблеме. Более подходящего слова, чем «прорыв» для описания результатов работы AlphaFold2 не нашлось ни у кого. А пресс-релиз DeepMind про победу на этом соревнований вызвал бурные обсуждения не только у профессионального сообщества и сочувствующих, но и у людей, далеких от переживаний по поводу предсказания структуры белков, биологии и всего, что с ними связано.
Здесь мы обсудим, что это за задача предсказания структуры и почему она такая сложная. Расскажем, почему она важна не только для академического сообщества, но и для того, что принято называть «индустрией». И, конечно, тоже выскажем свое мнение о том, что означает этот прорыв искусственного интеллекта для науки и индустрии в целом и для нас — простых людей, которые иногда болеют и лечатся от своих заболеваний, в частности.
Структура белка: что такое и зачем нужна
Так как Хабр — ресурс в первую очередь про IT и все, что с ним связано, для понимания всего того переполоха, который вызвал AlphaFold2, имеет смысл немного осветить предметную структурно-биологическую область.
Начнем с самого сложного — определения белка и описания его структуры. Белки, они же полипептиды, они же протеины — молекулы, обеспечивающие протекание большей части процессов в нашем организме. Они могут передавать и принимать сигналы, которыми обмениваются клетки нашего организма, могут участвовать в обмене веществ (как, например, инсулин, отвечающий за поддержание уровня глюкозы в организме). И даже антитела — те самые активные участники иммунного ответа на различные патогены — тоже являются белками. В общем белки всегда, белки везде. Но почему они такие могущественные и многопрофильные?
То, какую функцию выполняет белок и насколько хорошо он это делает, определяется его составом и структурой. Их аж целых 4 вида.
Первичная структура — так называемая последовательность белка. Белки состоят из 20 стандартных «строительных блоков», называемых альфа-аминокислотами. Каждая из аминокислот имеет обозначение в виде буквы: например, глицин — G, аланин — A, а аспарагин — N. Вот и получается, что для каждого белка мы можем записать строчку из 20-буквенного алфавита:
Insulin: MALWMRLLPLLALLALWGPDPAAAFVNQHLCGSHLVEALYLVCGERGFFYTPKTRREAEDLQVGQVELGGGPGAGSLQPLALEGSLQKRGIVEQCCTSICSLYQLENYCN
PD-1: MQIPQAPWPVVWAVLQLGWRPGWFLDSPDRPWNPPTFSPALLVVTEGDNATFTCSFSNTSESFVLNWYRMSPSNQTDKLAAFPEDRSQPGQDCRFRVTQLPNGRDFHMSVVRARRNDSGTYLCGAISLAPKAQIKESLRAELRVTERRAEVPTAHPSPSPRPAGQFQTLVVGVVGGLLGSLVLLVWVLAVICSRAARGTIGARRTGQPLKEDPSAVPVFSVDYGELDFQWREKTPEPPVPCVPEQTEYATIVFPSGMGTSSPARRGSADGPRSAQPLRPEDGHCSWPL
Зная последовательность ДНК (а точнее, РНК, можно однозначно определить эту самую последовательность букв для белков с помощью таблицы генетического кода:
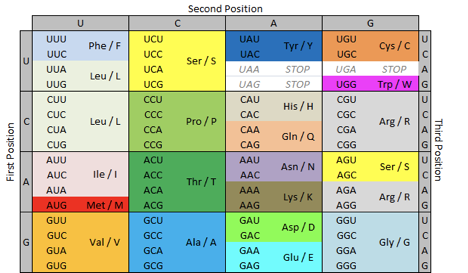
Таблица перевода троек (триплетов) нуклеотидов в аминоксилоты
Но букв, и даже строк, нам не достаточно. Чтобы понять, как работает белок, с чем и как в организме он способен взаимодействовать, нам нужно знать, как он устроен в пространстве.
Каждую аминокислоту можно представить как набор атомов, определенным образом расположенный в таком привычном нам трехмерном пространстве. У каждого атома — своя координата и свой набор связей с соседними атомами. Вот так, например, выглядит треонин:

Структура аминоксиолоты треонин в атомном представлении
Последовательности аминокислот способны укладываться в пространстве в стабильные вторичные структуры — так называемые альфа-спирали и бета-слои (и еще всякие разные), которые принято визуализировать изящными лентами:

Альфа-спираль

Бета-складка
А целый белок, состоящий из множества аминокислот, и, следовательно, из множества атомов со своими координатами, может выглядеть вот так:

То, как полноценная аминокислотная цепочка уложена в пространстве, называется третичной структурой. В ней зашито все: из каких остатков и атомов состоит белок и как они друг относительно друга расположены в трехмерном пространстве.
Есть еще более сложные белки — состоящие из нескольких полипептидных цепочек и способные функционировать только в таком виде. Структура таких белков в трехмерном пространстве называется четвертичной. Яркий представитель таких белков — гемоглобин, разносящий кислород по нашим тканям:

Структура гемоглобина
Именно полная пространственная структура белка (третичная или четвертичная) называется в обиходе просто структурой. И именно она полностью определяет его функцию. По трехмерной структуре белка становится понятно, какие аминокислоты способны образовывать взаимодействия с другими веществами, насколько сильными будут эти взаимодействия и к чему эти взаимодействия приведут: например, к расщеплению других белков или поддержанию правильной формы укладки молекул ДНК в клетке.
Зная структуру белка, можно рационально подойти к созданию лекарственного препарата, который должен с ним взаимодействовать. Или скорректировать крутой существующий белок из животных так, чтобы он не вызывал иммунного ответа у человека и лечить человека им. А если обладать возможностью по последовательности строить структуру белка, то можно даже сделать свой искусственный белок, который будет выполнять функцию, для которой не существует белка природного — например, расщеплять пластик. В общем, пространство для маневра не ограничено, а горизонты широки.
Именно поэтому над проблемой получения структуры белка бьются многие научные (и не только) группы. Традиционный подход к определению структуры белка — экспериментальный. Сделать это можно, например, с помощью рентгеноструктурного анализа (РСА), ядерного магнитного резонанса (ЯМР) или криоэлектронной микроскопии (Cryo-EM). У всех этих методов есть свои плюсы и минусы. Они предоставляют довольно надежную информацию о том, как же в пространстве организован белок, как атомы в его аминокислотах расположены друг относительно друга — все, что нужно. Но есть у них и три общих минуса: дорого, долго и сложно. Иногда просто невозможно получить образец белка в виде, нужном для проведения РСА. Расшифровать результаты ЯМР для белков длиннее 200 аминокислот — все еще нетривиальная задача. А хороший криоэлектронный микроскоп стоит несколько миллионов долларов, да и специалистов, владеющих искусством его укрощения, пока даже во всем мире не так много.
Это все к тому, что вопрос “А не предсказывать ли нам структуру белка по его последовательности биоинформатическими методами?” уже давно не подкупает новизной — попыток решить эту задачу было очень много (и, уверены мы, еще много будет!). Действительно успешными до 2020 года их назвать нельзя, и почему это так, мы поговорим ближе к концу статьи. Но история того, какие подходы применялись и насколько рабочими они были, не может идти в отрыве от истории соревнования под названием CASP.
CASP
Critical Assessment of protein Structure Prediction — или просто CASP — соревнование по предсказанию трехмерной структуры белков по их последовательности. Оно проходит раз в два года, и участвуют в нем все уважающие себя группы, разработавшие новый алгоритм для решения этой задачи. Это событие проходит с 1994 года, тогда в нем приняли участие 35 групп. А вот в 2020 уже больше сотни. В чем же они соревнуются?
Организаторы предлагают участникам предсказать структуры белков, зная лишь их последовательность. Эти структуры уже получены экспериментальными методами, перечисленными выше, но пока что их никто не видел. Обычно все разрешенные структуры публикуются в базе Protein Data Bank, и там их может найти любой желающий. Но некоторые экспериментаторы приберегают свои результаты как раз для такого случая.
Участвуя в CASP, можно показать класс в разных категориях предсказания структур — например, превзойти всех в качестве предсказания упаковки неупорядоченных структур или контактов между разными участками белка. Или еще попробовать предсказать третичную структуру белка по гомологии. Например, можно найти белок, наиболее похожий по последовательности на тот, структуру которого нужно предсказать, и координаты атомов для которого уже известны. И затем всяческими модификациями довести этот «шаблон» до красивой искомой третичной структуры требуемого белка. Как выбирается шаблон и к каким ухищрениям прибегают в этой категории, можно узнать здесь: Гомологичный фолдинг белков | Павел Яковлев (BIOCAD).

Но самой престижной и волнующей общественность категорией традиционно считается предсказание третичной структуры белка de novo (или ab initio). Эти красивые латинские слова означают, что при предсказании вы не используете напрямую известные структуры белков, а пользуетесь некими закономерностями и правилами для построения структуры с нуля.
При этом организаторов не очень волнует полная структура белков с расположениями всех атомов. Им интересны только так называемые Cα атомы — те, из которых растут радикалы аминокислот (на этом рисунке обозначены голубым цветом):

Тример аминокислот. Голубым обозначены Cα атомы (атомы водорода убраны для чистоты картинки)
«Как же так?!», возмущенно спросите вы. Мы тут 20 минут читали про атомы, про координаты, про то, как это важно для функции белка, и т.д. и т.п., а теперь давайте все выкинем и будем только вот этот несвязный набор точек предсказывать!
Сейчас станет понятно, почему так можно. Давайте сначала уберем эти самые радикалы и посмотрим, как белок выглядит без них:

Полипептидная цепочка без радикало аминокислот. Голубым обозначены Cα атомы (атомы водорода убраны для чистоты картинки)
В целом общая форма белка понятна. Восстановить направление того, что растет из Cα, можно опираясь на известные структуры из PDB (а их больше 170 тысяч): просто взять и по общему строению этого белка выбрать для него более подходящие ориентации радикалов.
А если убрать вообще все, кроме Cα, то выглядеть это будет так:

Расположение Cα атомов в белке
Выглядит плохо, но все не так безнадежно. Мы знаем, что между двумя Cα всегда находятся атомы углерода и азота. А еще благодаря квантовой механике мы знаем, какие расстояния связей между ними — они всегда одинаковые. Более того, с 1963 года [10.1016/S0022-2836(63)80023-6] мы даже знаем, каковы допустимые углы между плоскостями (они же двугранные углы), которые они образуют:

Определение углов phi и psi в белке

Карта Рамачандрана — распределение наблюдаемых значений phi и psi в белках в целом
Так что зная расстояния и углы между пропавшими атомами, восстановить их координаты — дело техники.
Итак, участники CASP по имеющейся аминокислотной последовательности белка предсказывают положение Cα атомов в пространстве. Теперь пора оценить, кто же из них лучший. Для этого есть специальная метрика — GDT_TS.
В конце соревнования у организаторов есть реальные положения Cα из эксперимента, и есть предсказанные участниками. Для оценки качества предсказаний сначала следует их совместить друг с другом, например, так:

Две совмещенные структуры одного белка
Делается это с помощью алгоритмов структурного выравнивания (например, вот такого). Теперь можно оценить схожесть полученных структур как раз по этой метрике. GDT(X) — Global Distance Test от X — это доля тех Cα, которые после структурного выравнивания находятся от референса не больше, чем на заданное расстояние X. На рисунке выше, например, один из предсказанных Cα находится на расстоянии 7.3 Å от референсного. Доля, как водится, распределена от 0 до 100%. GDT_TS — Global Distance Test Total Score в CASP определяется вот так:
GDT_TS = 1/4(GDT(1 Å) + GDT(2 Å) + GDT(4 Å) + GDT(8 Å))
Но это еще не все. Полученные потом и кровью участников показатели GDT_TS на всех предсказаниях структуры преобразуются в Z-score. При этом выкидываются выбросы и слишком низкие значения (с порогами -2 или 0) обрезаются.
Итоговые метрики для каждой команды (а их целых 4!) — суммарный Z-score для порога -2, суммарный Z-score для порога 0, средний Z-score для порога -2 и средний Z-score для порога 0.
Поговаривают, что это позволяет стать победителями, серебряными призерами и т.д. сразу нескольким командам. Но в 2020 году победитель по всем показателям был один — AlphaFold2. Цифра 2 в конце намекает, что у этого инструмента есть история. О ней сейчас и поговорим.
CASP13, CASP14 и DeepMind
В 2018 году состоялось соревнование CASP13, в котором, пусть и не разгромную, но очень убедительную победу одержала команда под названием A7D:
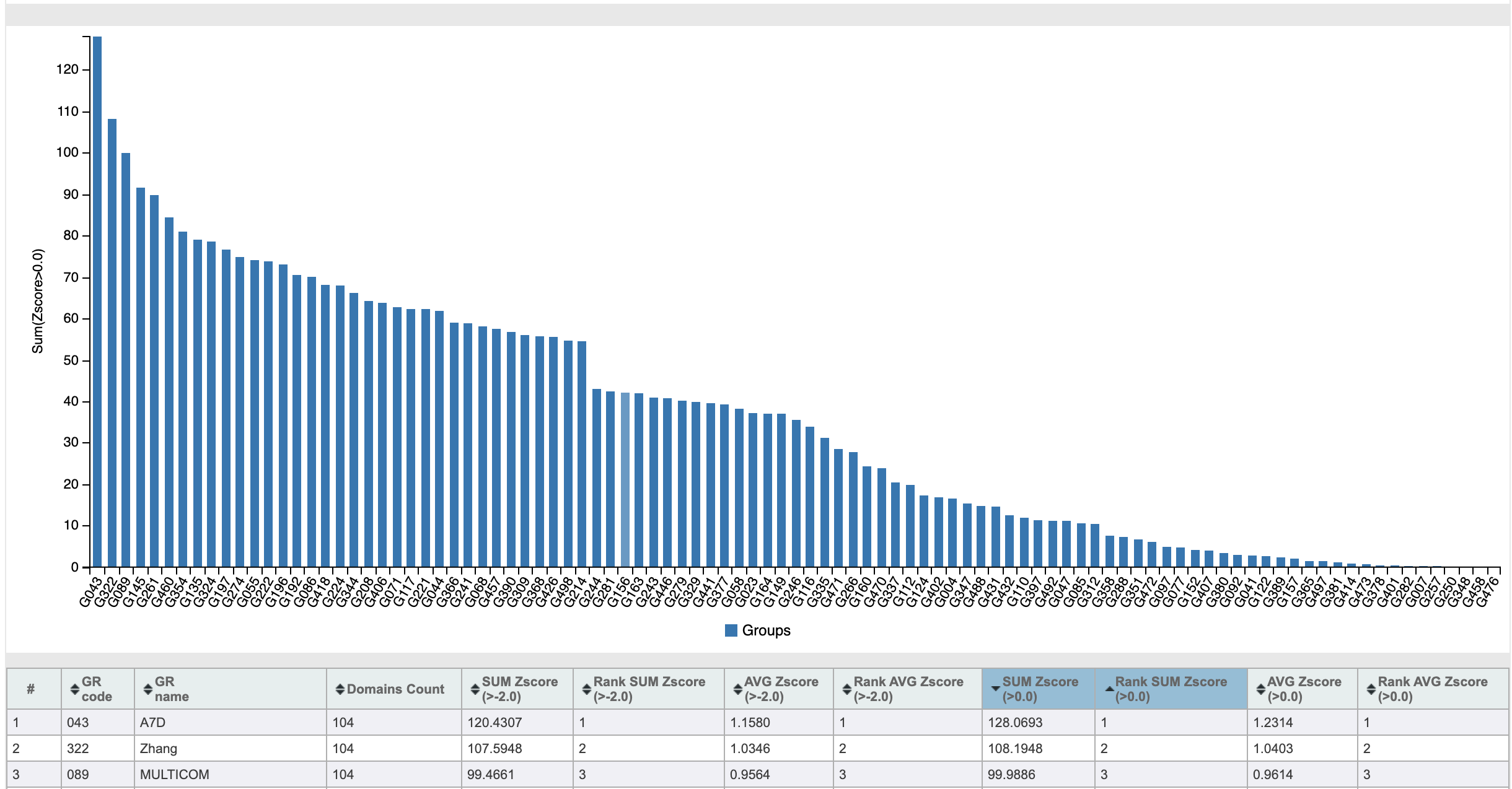
Показатели Z-score команд-участниц CASP13
Этим A7D был… нет, не Альберт Эйнштейн. А команда из компании DeepMind, разработавшая алгоритм AlphaFold. DeepMind известна всем, кто интересуется искусственным интеллектом. Основаны они были всего 10 лет назад, а в 2014 году были приобретены компанией Google. За недолгие 10 лет своего существования эти ребята успели отметиться во многих сферах. Искусственный интеллект AlphaGo в 2016 году победил чемпиона мира по игре го. Эта игра требует развитого стратегического и тактического мышления, присущего ранее только человеку. В 2019 году, после ряда успехов в создании искусственного интеллекта, способного играть в видеоигры не хуже людей, нейросеть AlphaStar за 44 дня стала гроссмейстером игры Starcraft II.
В 2020 году на CASP14 результаты выглядели уже вот так:

Таких чисел на CASP не видел никто. Еще более впечатляющим выглядит показатель, не зависящий от результатов других команд: меданный по всем структурам GDT_TS у AlphaFold2 составил 92.4. А это означает (по крайней мере, по пересчетам самого DeepMind), что их точность предсказания сопоставима с точностью разрешения структуры, которую дают экспериментальные методы. Но лучше всего качество предсказания структуры отражают, конечно, красивые и действительно впечатляющие картинки:

Источник: deepmind.com/blog/article/alphafold-a-solution-to-a-50-year-old-grand-challenge-in-biology
Бесспорно, результаты крутые. Давайте разберемся, что же такого скрыто под капотом AlphaFold и AlphaFold2, что позволяет им решать эту задачу с такой потрясающей точностью.
AlphaFold
Понятно, что результаты AlphaFold2 намного более впечатляющие, и интересно было бы разобраться, как работает именно он. Но, увы, пока у нас нет никакой подробной информации от его создателей — только пресс-релиз, посвященный победе в CASP14. Тем не менее подсказки о том, как он работает, можно найти в статье про первую версию AlphaFold.
Хотя победа на CASP13 состоялась в 2018 году, статья в престижном журнале «Природа», которая описывает алгоритм работы AlphaFold, вышла только в январе 2020 года. Так что публикацию про AlphaFold2 придется еще подождать. А пока поговорим про первую версию.
AlphaFold — сверхточная нейросеть. Соответственно, у нее есть фичи, принимаемые на вход, архитектура, через которую эти фичи проходят, и итоговые предсказанные данные, которые из этой нейросети выходят. Начнем с входных фичей.
Вход AlphaFold
Напомним, задача состоит в предсказании структуры белка по его аминокислотной последовательности. Но AlphaFold не так прост: на вход нейросеть принимает не только саму последовательность, а ее множественное выравнивание на последовательности из разных больших баз данных существующих белков.
И за этим стоит очень красивая идея, которую использовала команда DeepMind. Она прекрасно демонстрирует, что для решения задач биоинформатическими методами нужно быть не только хорошим математиком и программистом, но и понимать их биологический фундамент.
Итак, идея состоит в следующем. Мы берем последовательность белка и выравниваем ее на все известные человечеству последовательности белков. Их очень-очень много, намного больше, чем структур, ведь, как мы помним, получить последовательность белка, зная последовательность ДНК, очень просто. А проекты по секвенированию — определению последовательности ДНК для разных животных — за последние годы нагенерировали столько данных, что не использовать их было бы преступлением.
И вот, мы нашли похожие друг на друга по последовательности белки. Если они похожи по последовательности, то похожи они, скорее всего, и по функции, и по структуре. Но ведь не обязательно есть структура этих похожих белков (скорее всего, ее и нет). А нам она сейчас и не нужна, ведь и схожести мы можем вывести другие закономерности.
Аминокислоты белка между собой взаимодействуют — формируют устойчивые (и не очень) связи, которые формируют, поддерживают и стабилизируют его структуру.

И если одна из взаимодействующих аминокислот внезапно поменяется, сдвинется или пропадет, то ее визави может расстроиться, сместиться и перестать поддерживать структуру. Из-за этого белок расплетется и потеряет способность функционировать. А если он потеряет возможность нормально функционировать, может сильно пострадать здоровье и качество жизни организма, в котором он живет. И такой организм вряд ли долго проживет и размножится, и вряд ли мы успеем его просеквенировать.
Какой из этого можно сделать вывод? А такой, что если пары аминокислот образуют критические взаимодействия, то в похожих белках они либо не меняются, либо меняются синхронно. Поэтому, глядя на выравнивание похожих белков, можно посчитать корреляцию каждой пары позиций и построить то, что называется матрицей коэволюции. Где корреляция выше, там, вероятнее всего, есть критические взаимодействия, а, следовательно, эти позиции, вероятнее всего, находятся в пространстве рядом друг с другом.

Иллюстрация идеи использования информации о коэволюции для предсказания структуры Источник: predictioncenter.org/casp14/doc/presentations/2020_11_30_CASP14_Introduction_Moult.pdf
Идея хоть принадлежит и не DeepMind, показала себя она во все красе именно в их руках.
Выход AlphaFold
Поговорим теперь о том, что же AlphaFold выдает на выходе. Начнем, как обычно, издалека.
В задаче требуется предсказать положение всех Cα белка. Для решения этой проблемы можно заняться предсказанием координат атомов. Но представьте себе белок с одними координатами атомов. А теперь отнесите его на два метра вверх, один метр влево и 78 сантиметров вперед. И поверните вокруг центра масс на 15 градусов. Белок и структура остались теми же, а вот координаты сильно изменились. Поэтому предсказание трехмерных координат — дело неблагодарное, и так никто не делает. Так что надо придумать что-то другое. И прежде чем описать «другое», снова окунемся в биологическую составляющую задачи и узнаем, что такое атомы Cβ.
Cβ — атомы, связанные с Cα (обозначены розовым). Они есть у 19 аминокислот из 20. Глицин довольствуется только Cα.

Чем хорош Cβ, так это тем, что по его расстоянию до Cβ другой аминокислоты в белке можно понять, взаимодействуют ли эти остатки друг с другом. Если расстояние меньше 8 Å — взаимодействие есть, больше или равно — нет. Верно и обратное: если мы знаем, что взаимодействие между аминокислотными остатками есть, то и расстояние между их Cβ должно быть в итоговой структуре меньше 8 Å. Понимаете, куда мы клоним?
Команда AlphaFold придумала предсказывать попарные расстояния между атомами Cβ и на выходе нейросеть выдает дискретные плотности распределения вероятности попарных расстояний между этими атомами разных остатков. Для остатка под номером 29 это выглядит вот так:

Источник: www.nature.com/articles/s41586-019-1923-7
Распределение вероятностей расстояний для Cβ — это очень здорово, конечно, но нам тут нужна структура или хотя бы положение Cα. Не проблема — сейчас все будет.
Из этих вероятностей для каждой аминокислоты строится дистограмма — матрица предсказанных попарных расстояний между Cβ:

Источник: www.nature.com/articles/s41586-019-1923-7
Расстояния междe остатками мы знаем, можно и структуру восстановить. Для этого DeepMind сконструировали вот такой потенциал, зависящий от углов фи и пси — тех самых двугранных углов, про распределение значений которых мы говорили ранее:

Первый вклад представляет из себя потенциал, вносимый предсказанным расположением Cb — G(phi, psi). Это функция, выражающая их попарное расстояние через эти углы. Второй вклад пришел к нам из небольшого сюрприза, который можно выявить, только если очень внимательно прочитать статью. Вместе с попарными расстояниями DeepMind предсказывают и распределение вероятностей значений двугранных углов фи и пси — отсюда и вклад в потенциал. А третий вклад предназначен для того, чтобы избежать ситуации, когда все построенные атомы друг с другом сталкиваются и их координаты перекрываются.
Этот потенциал — ни что иное, как математическая модель, описывающая потенциальную энергию этого белка. Таких моделей много (вот тут можно посмотреть, какие они есть), но команда AlphaFold создала свою. Законы термодинамики говорят нам, что закрытая система стремится к минимуму потенциальной энергии. И если мы знаем функциональный вид этой энергии, и, более того, он еще и дифференцируемый, как в этом случае, то можно минимизировать его и прийти к реальной структуре. Так что на этом этапе возникает вполне себе сухая математическая задача оптимизации функционала. Для ее решения команда использует метод градиентного спуска.
И в итоге имеются значения всех фи и пси из оптимальной структуры. Расстояние между Cα, C и N мы знаем. Восстановить из этого корректные относительные положения Cα не представляет никакой трудности.
Итак, со входными фичами все понятно. С постобработкой результатов алгоритма — тоже. А что сидит внутри этой нейросети, которая столь точно предсказывает попарные расстояния Cβ и распределение двугранных углов?
Архитектура AlphaFold
В статье приведена схема нейросети:

Это сверточная нейросеть, которая относится к классу ResNet, что позволяет ей обучаться более эффективно. Обучающей выборкой служили ~30 000 экспериментально полученных структур из базы PDB, про которую мы с вами говорили выше, и обучение заняло примерно 5 дней.
Вообще говоря, какого-то прорыва именно в построении архитектуры сети здесь не случилось — просто мощная и логично выстроенная нейросеть. И успех ее кроется, конечно, именно в выборе входных и выходных данных.
AlphaFold2
А что же AlphaFold2? Про его устройство мы знаем не так много. Схематично его архитектура выглядит вот так:

Из пресс-релиза мы знаем, что эта нейросеть — 'end-to-end', то есть (наверное) ей достаточно на вход иметь только последовательность белка, на основании которой она самостоятельно сделает множественное выравнивание и вытащит из него нужные фичи. И на выходе нас сразу будет ждать структура.
В этот раз обучение проходило на примерно 170 000 структур из PDB, и заняло оно несколько недель.
И, кажется, идея все такая же — из данных о корреляции позиций получить распределение вероятностей попарных расстояний. Или нет. В общем, будем с нетерпением ждать статью в престижном журнале.
Discussion
В отличие от парламента, пост на Хабре и комментарии к нему — вполне уютное место для дискуссии, поэтому здесь мы выскажем несколько мнений по поводу всех вышеизложенных фактов. И начать хочется с разбора того, что вызвало общее возмущение у причастных к структурной и вычислительной биологии.
Анонс DeppMind, на который мы так часто здесь ссылались, называется «AlphaFold: a solution to a 50-year-old grand challenge in biology». И в первом же абзаце там говорится о том, что этой команде удалось решить проблему фолдинга. Читая это, многие взрывались, причем по разным причинам.
Во-первых, строго говоря, никакую задачу фолдинга они не то что не решили, они ее и не решали. Фолдинг — это процесс приобретения белком его третичной структуры. То есть решение задачи фолдинга — это выявление закономерностей, приводящих последовательность белка к различным этапам формирования его структуры и к окончательной третичной структуре. И задача эта куда более сложная, ведь белки пришивают к себе по одной аминокислоте и начинают формировать свою структуру по мере своего роста в сложной многокомпонентной среде. А еще делать это они могут по-разному в зависимости от внешних условий. И путей прихода к конечной структуре у них бесконечное множество.
При этом известно, что белки не занимаются просто перебором всех возможных конформаций. Парадокс Левинталя как раз говорит о том, что если бы белок из 100 аминокислот поступал именно так, то даже при дикой скорости перебора конформаций времени жизни Вселенной для приобретения своей структуры ему бы не хватило. Основная теория, которая сейчас превалирует на полях решения задачи фолдинга, называется догмой Анфинсена и гласит, что белок последовательно идет к кинетически достижимому энергетическом минимуму.
Очевидно, что задача предсказания процесса формирования структуры намного сложнее, чем то, что делали DeepMind, ведь они решали задачу предсказания структуры. И такая подмена понятий вызвала в чем-то справедливое негодование сочувствующих процессу граждан.
Справедливости ради стоит отметить пару моментов. Во-первых, термин фолдинг очень часто употребляется вычислительными биологами именно в значении предсказание структуры. Правда, это делается в узких кругах, и в приличном обществе вслух такое не говорят. Так что можно считать, что пресс-релиз DeepMind просто содержал не к месту употребленный жаргонизм. Во-вторых, если прочитать этот релиз чуть дальше первого абзаца, его авторы как раз поясняют, что решали они именно задачу предсказания структуры, без всяких там процессов. Ну, это все демагогия и борьба за чистоту языка. Давайте перейдем к во-вторых и к размышлениям по делу.
А именно к размышлениям о слове «решение». Когда речь идет об использовании нейросетей для определения лиц или подсчета количества котиков на картинке, мы всегда можем понять, ошиблась нейросеть или нет. Просто потому что большинство людей способны по картинке определить, сколько на ней котиков и тот ли на ней человек, которого они перед собой видят. А вот когда мы используем нейросеть для решения задачи, ответа на которую не знаем, возникают вопросы. Как мы поймем, что нейросеть сработала верно и не сломалась? С какой долей уверенности мы можем использовать результаты работы этой нейросети в своей практической деятельности? Например, в проекте по разработке лекарственных препаратов, провал которого стоит несколько миллионов (иногда даже долларов)? Пока у нас нет ответа на этот вопрос, следовательно, говорить о решении, наверное, рановато.
Конечно, и в первой, и во второй версиях AlphaFold упоминается скор, призванный отличить хорошую работу алгоритма от плохой. Но вот данных по тому, насколько он соотносится с реальностью в открытом доступе, к сожалению, нет. Так что о практическом и рутинном применении речи, увы, пока не идет.
И тем не менее. Нельзя не отметить, что этот алгоритм показал работу, значительно превосходящую по результату все предыдущие попытки решить задачу предсказания структуры белка. И это означает, что потенциал метода, лежащего в основе этого алгоритма — огромный, и применять подобный метод можно не только к этой наукоемкой и сложной задаче. А значит, при должном развитии таких подходов нас ждет очень светлое и увлекательное будущее!
Заключение
В заключение хочется сказать следующее — AlphaFold2, безусловно, прорыв. Пока что есть некоторые моменты, ограничивающие его практическое применение, но, кажется, их можно преодолеть. Разумеется, для этого нужно задаться такой целью и уверенно к ней идти. И хочется верить, что DeepMind не бросит свое детище и будет и дальше его развивать и улучшать. Будет очень здорово, если это развитие будет проходить такими же семимильными шагами, как до этого.
Постзаключение
Надеемся, вас впечатлили существующие задачи, подходы к их решению и успехи этих подходов. На наш взгляд, самое красивое в таких событиях — это демонстрация того, как правильно примененные знания из разных областей могут дать невероятные результаты и помочь в решении сложнейших задач, стоящих перед человечеством.
AlphaFold появился на свет благодаря тому, что члены его команды обладали компетенциями в области биологии, физики, математики, алгоритмов глубокого обучения и оптимизации — то есть в области вычислительной биологии. Этому мало где хорошо учат в мире и пока что нигде на достаточном уровне не учат в России. Но в 2021 году в Высшая Школа Экономики совместно с компанией BIOCAD запускает магистерскую программу «Вычислительная биология и биоинформатика», в которой будут учить этим дисциплинам, столь необходимым для решения таких амбициозных задач.
В магистратуре ждут студентов с сильным физико-математическим бэкграундом, без химической и биологической подготовки. Преподаватели ВШЭ обеспечат лучшие в стране курсы по алгоритмам, программированию, анализу данных, а сотрудники индустрии расскажут спецглавы физики, молекулярной биологии, химии, а также специальные курсы по молекулярному моделированию, алгоритмам структурной биоинформатики, системной фармакологии и иным важным для области темам. И, что тоже важно, расскажут о настоящих индустриальных биологических задачах и научат использовать полученные знания и навыки для их решения.
Введение
Если вы когда-то слышали про биоинформатику, вычислительную биологию, структуру белка и проблему фолдинга, то вы знаете, о чем мы здесь будем рассказывать.
Если вы интересуетесь глубоким обучением и нейросетями, их применением для решения насущных практических задач и глубоко убеждены, что искусственный интеллект если и не завладеет миром, то точно превзойдет человека в своих когнитивных способностях, то об этой его победе над человеческим познанием вы тоже точно слышали.
Речь в нашей статье пойдет, как следует из названия, об искусственном интеллекте AlphaFold и его продолжателе AlphaFold2 от компании DeepMind, который был создан для предсказания трехмерной структуры белка. В 2020 году AlphaFold2 выполнил свою задачу так хорошо, как не удавалось никому до него, и с разгромными показателями победил своих соперников в соревновании, посвященном этой проблеме. Более подходящего слова, чем «прорыв» для описания результатов работы AlphaFold2 не нашлось ни у кого. А пресс-релиз DeepMind про победу на этом соревнований вызвал бурные обсуждения не только у профессионального сообщества и сочувствующих, но и у людей, далеких от переживаний по поводу предсказания структуры белков, биологии и всего, что с ними связано.
Здесь мы обсудим, что это за задача предсказания структуры и почему она такая сложная. Расскажем, почему она важна не только для академического сообщества, но и для того, что принято называть «индустрией». И, конечно, тоже выскажем свое мнение о том, что означает этот прорыв искусственного интеллекта для науки и индустрии в целом и для нас — простых людей, которые иногда болеют и лечатся от своих заболеваний, в частности.
Структура белка: что такое и зачем нужна
Так как Хабр — ресурс в первую очередь про IT и все, что с ним связано, для понимания всего того переполоха, который вызвал AlphaFold2, имеет смысл немного осветить предметную структурно-биологическую область.
Начнем с самого сложного — определения белка и описания его структуры. Белки, они же полипептиды, они же протеины — молекулы, обеспечивающие протекание большей части процессов в нашем организме. Они могут передавать и принимать сигналы, которыми обмениваются клетки нашего организма, могут участвовать в обмене веществ (как, например, инсулин, отвечающий за поддержание уровня глюкозы в организме). И даже антитела — те самые активные участники иммунного ответа на различные патогены — тоже являются белками. В общем белки всегда, белки везде. Но почему они такие могущественные и многопрофильные?
То, какую функцию выполняет белок и насколько хорошо он это делает, определяется его составом и структурой. Их аж целых 4 вида.
Первичная структура — так называемая последовательность белка. Белки состоят из 20 стандартных «строительных блоков», называемых альфа-аминокислотами. Каждая из аминокислот имеет обозначение в виде буквы: например, глицин — G, аланин — A, а аспарагин — N. Вот и получается, что для каждого белка мы можем записать строчку из 20-буквенного алфавита:
Insulin: MALWMRLLPLLALLALWGPDPAAAFVNQHLCGSHLVEALYLVCGERGFFYTPKTRREAEDLQVGQVELGGGPGAGSLQPLALEGSLQKRGIVEQCCTSICSLYQLENYCN
PD-1: MQIPQAPWPVVWAVLQLGWRPGWFLDSPDRPWNPPTFSPALLVVTEGDNATFTCSFSNTSESFVLNWYRMSPSNQTDKLAAFPEDRSQPGQDCRFRVTQLPNGRDFHMSVVRARRNDSGTYLCGAISLAPKAQIKESLRAELRVTERRAEVPTAHPSPSPRPAGQFQTLVVGVVGGLLGSLVLLVWVLAVICSRAARGTIGARRTGQPLKEDPSAVPVFSVDYGELDFQWREKTPEPPVPCVPEQTEYATIVFPSGMGTSSPARRGSADGPRSAQPLRPEDGHCSWPL
Зная последовательность ДНК (а точнее, РНК, можно однозначно определить эту самую последовательность букв для белков с помощью таблицы генетического кода:
Таблица перевода троек (триплетов) нуклеотидов в аминоксилоты
Но букв, и даже строк, нам не достаточно. Чтобы понять, как работает белок, с чем и как в организме он способен взаимодействовать, нам нужно знать, как он устроен в пространстве.
Каждую аминокислоту можно представить как набор атомов, определенным образом расположенный в таком привычном нам трехмерном пространстве. У каждого атома — своя координата и свой набор связей с соседними атомами. Вот так, например, выглядит треонин:
Структура аминоксиолоты треонин в атомном представлении
Последовательности аминокислот способны укладываться в пространстве в стабильные вторичные структуры — так называемые альфа-спирали и бета-слои (и еще всякие разные), которые принято визуализировать изящными лентами:
Альфа-спираль
Бета-складка
А целый белок, состоящий из множества аминокислот, и, следовательно, из множества атомов со своими координатами, может выглядеть вот так:
То, как полноценная аминокислотная цепочка уложена в пространстве, называется третичной структурой. В ней зашито все: из каких остатков и атомов состоит белок и как они друг относительно друга расположены в трехмерном пространстве.
Есть еще более сложные белки — состоящие из нескольких полипептидных цепочек и способные функционировать только в таком виде. Структура таких белков в трехмерном пространстве называется четвертичной. Яркий представитель таких белков — гемоглобин, разносящий кислород по нашим тканям:
Структура гемоглобина
Именно полная пространственная структура белка (третичная или четвертичная) называется в обиходе просто структурой. И именно она полностью определяет его функцию. По трехмерной структуре белка становится понятно, какие аминокислоты способны образовывать взаимодействия с другими веществами, насколько сильными будут эти взаимодействия и к чему эти взаимодействия приведут: например, к расщеплению других белков или поддержанию правильной формы укладки молекул ДНК в клетке.
Зная структуру белка, можно рационально подойти к созданию лекарственного препарата, который должен с ним взаимодействовать. Или скорректировать крутой существующий белок из животных так, чтобы он не вызывал иммунного ответа у человека и лечить человека им. А если обладать возможностью по последовательности строить структуру белка, то можно даже сделать свой искусственный белок, который будет выполнять функцию, для которой не существует белка природного — например, расщеплять пластик. В общем, пространство для маневра не ограничено, а горизонты широки.
Именно поэтому над проблемой получения структуры белка бьются многие научные (и не только) группы. Традиционный подход к определению структуры белка — экспериментальный. Сделать это можно, например, с помощью рентгеноструктурного анализа (РСА), ядерного магнитного резонанса (ЯМР) или криоэлектронной микроскопии (Cryo-EM). У всех этих методов есть свои плюсы и минусы. Они предоставляют довольно надежную информацию о том, как же в пространстве организован белок, как атомы в его аминокислотах расположены друг относительно друга — все, что нужно. Но есть у них и три общих минуса: дорого, долго и сложно. Иногда просто невозможно получить образец белка в виде, нужном для проведения РСА. Расшифровать результаты ЯМР для белков длиннее 200 аминокислот — все еще нетривиальная задача. А хороший криоэлектронный микроскоп стоит несколько миллионов долларов, да и специалистов, владеющих искусством его укрощения, пока даже во всем мире не так много.
Это все к тому, что вопрос “А не предсказывать ли нам структуру белка по его последовательности биоинформатическими методами?” уже давно не подкупает новизной — попыток решить эту задачу было очень много (и, уверены мы, еще много будет!). Действительно успешными до 2020 года их назвать нельзя, и почему это так, мы поговорим ближе к концу статьи. Но история того, какие подходы применялись и насколько рабочими они были, не может идти в отрыве от истории соревнования под названием CASP.
CASP
Critical Assessment of protein Structure Prediction — или просто CASP — соревнование по предсказанию трехмерной структуры белков по их последовательности. Оно проходит раз в два года, и участвуют в нем все уважающие себя группы, разработавшие новый алгоритм для решения этой задачи. Это событие проходит с 1994 года, тогда в нем приняли участие 35 групп. А вот в 2020 уже больше сотни. В чем же они соревнуются?
Организаторы предлагают участникам предсказать структуры белков, зная лишь их последовательность. Эти структуры уже получены экспериментальными методами, перечисленными выше, но пока что их никто не видел. Обычно все разрешенные структуры публикуются в базе Protein Data Bank, и там их может найти любой желающий. Но некоторые экспериментаторы приберегают свои результаты как раз для такого случая.
Участвуя в CASP, можно показать класс в разных категориях предсказания структур — например, превзойти всех в качестве предсказания упаковки неупорядоченных структур или контактов между разными участками белка. Или еще попробовать предсказать третичную структуру белка по гомологии. Например, можно найти белок, наиболее похожий по последовательности на тот, структуру которого нужно предсказать, и координаты атомов для которого уже известны. И затем всяческими модификациями довести этот «шаблон» до красивой искомой третичной структуры требуемого белка. Как выбирается шаблон и к каким ухищрениям прибегают в этой категории, можно узнать здесь: Гомологичный фолдинг белков | Павел Яковлев (BIOCAD).
Но самой престижной и волнующей общественность категорией традиционно считается предсказание третичной структуры белка de novo (или ab initio). Эти красивые латинские слова означают, что при предсказании вы не используете напрямую известные структуры белков, а пользуетесь некими закономерностями и правилами для построения структуры с нуля.
При этом организаторов не очень волнует полная структура белков с расположениями всех атомов. Им интересны только так называемые Cα атомы — те, из которых растут радикалы аминокислот (на этом рисунке обозначены голубым цветом):
Тример аминокислот. Голубым обозначены Cα атомы (атомы водорода убраны для чистоты картинки)
«Как же так?!», возмущенно спросите вы. Мы тут 20 минут читали про атомы, про координаты, про то, как это важно для функции белка, и т.д. и т.п., а теперь давайте все выкинем и будем только вот этот несвязный набор точек предсказывать!
Сейчас станет понятно, почему так можно. Давайте сначала уберем эти самые радикалы и посмотрим, как белок выглядит без них:
Полипептидная цепочка без радикало аминокислот. Голубым обозначены Cα атомы (атомы водорода убраны для чистоты картинки)
В целом общая форма белка понятна. Восстановить направление того, что растет из Cα, можно опираясь на известные структуры из PDB (а их больше 170 тысяч): просто взять и по общему строению этого белка выбрать для него более подходящие ориентации радикалов.
А если убрать вообще все, кроме Cα, то выглядеть это будет так:
Расположение Cα атомов в белке
Выглядит плохо, но все не так безнадежно. Мы знаем, что между двумя Cα всегда находятся атомы углерода и азота. А еще благодаря квантовой механике мы знаем, какие расстояния связей между ними — они всегда одинаковые. Более того, с 1963 года [10.1016/S0022-2836(63)80023-6] мы даже знаем, каковы допустимые углы между плоскостями (они же двугранные углы), которые они образуют:
Определение углов phi и psi в белке
Карта Рамачандрана — распределение наблюдаемых значений phi и psi в белках в целом
Так что зная расстояния и углы между пропавшими атомами, восстановить их координаты — дело техники.
Итак, участники CASP по имеющейся аминокислотной последовательности белка предсказывают положение Cα атомов в пространстве. Теперь пора оценить, кто же из них лучший. Для этого есть специальная метрика — GDT_TS.
В конце соревнования у организаторов есть реальные положения Cα из эксперимента, и есть предсказанные участниками. Для оценки качества предсказаний сначала следует их совместить друг с другом, например, так:
Две совмещенные структуры одного белка
Делается это с помощью алгоритмов структурного выравнивания (например, вот такого). Теперь можно оценить схожесть полученных структур как раз по этой метрике. GDT(X) — Global Distance Test от X — это доля тех Cα, которые после структурного выравнивания находятся от референса не больше, чем на заданное расстояние X. На рисунке выше, например, один из предсказанных Cα находится на расстоянии 7.3 Å от референсного. Доля, как водится, распределена от 0 до 100%. GDT_TS — Global Distance Test Total Score в CASP определяется вот так:
GDT_TS = 1/4(GDT(1 Å) + GDT(2 Å) + GDT(4 Å) + GDT(8 Å))
Но это еще не все. Полученные потом и кровью участников показатели GDT_TS на всех предсказаниях структуры преобразуются в Z-score. При этом выкидываются выбросы и слишком низкие значения (с порогами -2 или 0) обрезаются.
Итоговые метрики для каждой команды (а их целых 4!) — суммарный Z-score для порога -2, суммарный Z-score для порога 0, средний Z-score для порога -2 и средний Z-score для порога 0.
Поговаривают, что это позволяет стать победителями, серебряными призерами и т.д. сразу нескольким командам. Но в 2020 году победитель по всем показателям был один — AlphaFold2. Цифра 2 в конце намекает, что у этого инструмента есть история. О ней сейчас и поговорим.
CASP13, CASP14 и DeepMind
В 2018 году состоялось соревнование CASP13, в котором, пусть и не разгромную, но очень убедительную победу одержала команда под названием A7D:
Показатели Z-score команд-участниц CASP13
Этим A7D был… нет, не Альберт Эйнштейн. А команда из компании DeepMind, разработавшая алгоритм AlphaFold. DeepMind известна всем, кто интересуется искусственным интеллектом. Основаны они были всего 10 лет назад, а в 2014 году были приобретены компанией Google. За недолгие 10 лет своего существования эти ребята успели отметиться во многих сферах. Искусственный интеллект AlphaGo в 2016 году победил чемпиона мира по игре го. Эта игра требует развитого стратегического и тактического мышления, присущего ранее только человеку. В 2019 году, после ряда успехов в создании искусственного интеллекта, способного играть в видеоигры не хуже людей, нейросеть AlphaStar за 44 дня стала гроссмейстером игры Starcraft II.
В 2020 году на CASP14 результаты выглядели уже вот так:
Таких чисел на CASP не видел никто. Еще более впечатляющим выглядит показатель, не зависящий от результатов других команд: меданный по всем структурам GDT_TS у AlphaFold2 составил 92.4. А это означает (по крайней мере, по пересчетам самого DeepMind), что их точность предсказания сопоставима с точностью разрешения структуры, которую дают экспериментальные методы. Но лучше всего качество предсказания структуры отражают, конечно, красивые и действительно впечатляющие картинки:
Источник: deepmind.com/blog/article/alphafold-a-solution-to-a-50-year-old-grand-challenge-in-biology
Бесспорно, результаты крутые. Давайте разберемся, что же такого скрыто под капотом AlphaFold и AlphaFold2, что позволяет им решать эту задачу с такой потрясающей точностью.
AlphaFold
Понятно, что результаты AlphaFold2 намного более впечатляющие, и интересно было бы разобраться, как работает именно он. Но, увы, пока у нас нет никакой подробной информации от его создателей — только пресс-релиз, посвященный победе в CASP14. Тем не менее подсказки о том, как он работает, можно найти в статье про первую версию AlphaFold.
Хотя победа на CASP13 состоялась в 2018 году, статья в престижном журнале «Природа», которая описывает алгоритм работы AlphaFold, вышла только в январе 2020 года. Так что публикацию про AlphaFold2 придется еще подождать. А пока поговорим про первую версию.
AlphaFold — сверхточная нейросеть. Соответственно, у нее есть фичи, принимаемые на вход, архитектура, через которую эти фичи проходят, и итоговые предсказанные данные, которые из этой нейросети выходят. Начнем с входных фичей.
Вход AlphaFold
Напомним, задача состоит в предсказании структуры белка по его аминокислотной последовательности. Но AlphaFold не так прост: на вход нейросеть принимает не только саму последовательность, а ее множественное выравнивание на последовательности из разных больших баз данных существующих белков.
И за этим стоит очень красивая идея, которую использовала команда DeepMind. Она прекрасно демонстрирует, что для решения задач биоинформатическими методами нужно быть не только хорошим математиком и программистом, но и понимать их биологический фундамент.
Итак, идея состоит в следующем. Мы берем последовательность белка и выравниваем ее на все известные человечеству последовательности белков. Их очень-очень много, намного больше, чем структур, ведь, как мы помним, получить последовательность белка, зная последовательность ДНК, очень просто. А проекты по секвенированию — определению последовательности ДНК для разных животных — за последние годы нагенерировали столько данных, что не использовать их было бы преступлением.
И вот, мы нашли похожие друг на друга по последовательности белки. Если они похожи по последовательности, то похожи они, скорее всего, и по функции, и по структуре. Но ведь не обязательно есть структура этих похожих белков (скорее всего, ее и нет). А нам она сейчас и не нужна, ведь и схожести мы можем вывести другие закономерности.
Аминокислоты белка между собой взаимодействуют — формируют устойчивые (и не очень) связи, которые формируют, поддерживают и стабилизируют его структуру.
И если одна из взаимодействующих аминокислот внезапно поменяется, сдвинется или пропадет, то ее визави может расстроиться, сместиться и перестать поддерживать структуру. Из-за этого белок расплетется и потеряет способность функционировать. А если он потеряет возможность нормально функционировать, может сильно пострадать здоровье и качество жизни организма, в котором он живет. И такой организм вряд ли долго проживет и размножится, и вряд ли мы успеем его просеквенировать.
Какой из этого можно сделать вывод? А такой, что если пары аминокислот образуют критические взаимодействия, то в похожих белках они либо не меняются, либо меняются синхронно. Поэтому, глядя на выравнивание похожих белков, можно посчитать корреляцию каждой пары позиций и построить то, что называется матрицей коэволюции. Где корреляция выше, там, вероятнее всего, есть критические взаимодействия, а, следовательно, эти позиции, вероятнее всего, находятся в пространстве рядом друг с другом.
Иллюстрация идеи использования информации о коэволюции для предсказания структуры Источник: predictioncenter.org/casp14/doc/presentations/2020_11_30_CASP14_Introduction_Moult.pdf
Идея хоть принадлежит и не DeepMind, показала себя она во все красе именно в их руках.
Выход AlphaFold
Поговорим теперь о том, что же AlphaFold выдает на выходе. Начнем, как обычно, издалека.
В задаче требуется предсказать положение всех Cα белка. Для решения этой проблемы можно заняться предсказанием координат атомов. Но представьте себе белок с одними координатами атомов. А теперь отнесите его на два метра вверх, один метр влево и 78 сантиметров вперед. И поверните вокруг центра масс на 15 градусов. Белок и структура остались теми же, а вот координаты сильно изменились. Поэтому предсказание трехмерных координат — дело неблагодарное, и так никто не делает. Так что надо придумать что-то другое. И прежде чем описать «другое», снова окунемся в биологическую составляющую задачи и узнаем, что такое атомы Cβ.
Cβ — атомы, связанные с Cα (обозначены розовым). Они есть у 19 аминокислот из 20. Глицин довольствуется только Cα.
Чем хорош Cβ, так это тем, что по его расстоянию до Cβ другой аминокислоты в белке можно понять, взаимодействуют ли эти остатки друг с другом. Если расстояние меньше 8 Å — взаимодействие есть, больше или равно — нет. Верно и обратное: если мы знаем, что взаимодействие между аминокислотными остатками есть, то и расстояние между их Cβ должно быть в итоговой структуре меньше 8 Å. Понимаете, куда мы клоним?
Команда AlphaFold придумала предсказывать попарные расстояния между атомами Cβ и на выходе нейросеть выдает дискретные плотности распределения вероятности попарных расстояний между этими атомами разных остатков. Для остатка под номером 29 это выглядит вот так:
Источник: www.nature.com/articles/s41586-019-1923-7
Распределение вероятностей расстояний для Cβ — это очень здорово, конечно, но нам тут нужна структура или хотя бы положение Cα. Не проблема — сейчас все будет.
Из этих вероятностей для каждой аминокислоты строится дистограмма — матрица предсказанных попарных расстояний между Cβ:
Источник: www.nature.com/articles/s41586-019-1923-7
Расстояния междe остатками мы знаем, можно и структуру восстановить. Для этого DeepMind сконструировали вот такой потенциал, зависящий от углов фи и пси — тех самых двугранных углов, про распределение значений которых мы говорили ранее:
Первый вклад представляет из себя потенциал, вносимый предсказанным расположением Cb — G(phi, psi). Это функция, выражающая их попарное расстояние через эти углы. Второй вклад пришел к нам из небольшого сюрприза, который можно выявить, только если очень внимательно прочитать статью. Вместе с попарными расстояниями DeepMind предсказывают и распределение вероятностей значений двугранных углов фи и пси — отсюда и вклад в потенциал. А третий вклад предназначен для того, чтобы избежать ситуации, когда все построенные атомы друг с другом сталкиваются и их координаты перекрываются.
Этот потенциал — ни что иное, как математическая модель, описывающая потенциальную энергию этого белка. Таких моделей много (вот тут можно посмотреть, какие они есть), но команда AlphaFold создала свою. Законы термодинамики говорят нам, что закрытая система стремится к минимуму потенциальной энергии. И если мы знаем функциональный вид этой энергии, и, более того, он еще и дифференцируемый, как в этом случае, то можно минимизировать его и прийти к реальной структуре. Так что на этом этапе возникает вполне себе сухая математическая задача оптимизации функционала. Для ее решения команда использует метод градиентного спуска.
И в итоге имеются значения всех фи и пси из оптимальной структуры. Расстояние между Cα, C и N мы знаем. Восстановить из этого корректные относительные положения Cα не представляет никакой трудности.
Итак, со входными фичами все понятно. С постобработкой результатов алгоритма — тоже. А что сидит внутри этой нейросети, которая столь точно предсказывает попарные расстояния Cβ и распределение двугранных углов?
Архитектура AlphaFold
В статье приведена схема нейросети:
Это сверточная нейросеть, которая относится к классу ResNet, что позволяет ей обучаться более эффективно. Обучающей выборкой служили ~30 000 экспериментально полученных структур из базы PDB, про которую мы с вами говорили выше, и обучение заняло примерно 5 дней.
Вообще говоря, какого-то прорыва именно в построении архитектуры сети здесь не случилось — просто мощная и логично выстроенная нейросеть. И успех ее кроется, конечно, именно в выборе входных и выходных данных.
AlphaFold2
А что же AlphaFold2? Про его устройство мы знаем не так много. Схематично его архитектура выглядит вот так:
Из пресс-релиза мы знаем, что эта нейросеть — 'end-to-end', то есть (наверное) ей достаточно на вход иметь только последовательность белка, на основании которой она самостоятельно сделает множественное выравнивание и вытащит из него нужные фичи. И на выходе нас сразу будет ждать структура.
В этот раз обучение проходило на примерно 170 000 структур из PDB, и заняло оно несколько недель.
И, кажется, идея все такая же — из данных о корреляции позиций получить распределение вероятностей попарных расстояний. Или нет. В общем, будем с нетерпением ждать статью в престижном журнале.
Discussion
В отличие от парламента, пост на Хабре и комментарии к нему — вполне уютное место для дискуссии, поэтому здесь мы выскажем несколько мнений по поводу всех вышеизложенных фактов. И начать хочется с разбора того, что вызвало общее возмущение у причастных к структурной и вычислительной биологии.
Анонс DeppMind, на который мы так часто здесь ссылались, называется «AlphaFold: a solution to a 50-year-old grand challenge in biology». И в первом же абзаце там говорится о том, что этой команде удалось решить проблему фолдинга. Читая это, многие взрывались, причем по разным причинам.
Во-первых, строго говоря, никакую задачу фолдинга они не то что не решили, они ее и не решали. Фолдинг — это процесс приобретения белком его третичной структуры. То есть решение задачи фолдинга — это выявление закономерностей, приводящих последовательность белка к различным этапам формирования его структуры и к окончательной третичной структуре. И задача эта куда более сложная, ведь белки пришивают к себе по одной аминокислоте и начинают формировать свою структуру по мере своего роста в сложной многокомпонентной среде. А еще делать это они могут по-разному в зависимости от внешних условий. И путей прихода к конечной структуре у них бесконечное множество.
При этом известно, что белки не занимаются просто перебором всех возможных конформаций. Парадокс Левинталя как раз говорит о том, что если бы белок из 100 аминокислот поступал именно так, то даже при дикой скорости перебора конформаций времени жизни Вселенной для приобретения своей структуры ему бы не хватило. Основная теория, которая сейчас превалирует на полях решения задачи фолдинга, называется догмой Анфинсена и гласит, что белок последовательно идет к кинетически достижимому энергетическом минимуму.
Очевидно, что задача предсказания процесса формирования структуры намного сложнее, чем то, что делали DeepMind, ведь они решали задачу предсказания структуры. И такая подмена понятий вызвала в чем-то справедливое негодование сочувствующих процессу граждан.
Справедливости ради стоит отметить пару моментов. Во-первых, термин фолдинг очень часто употребляется вычислительными биологами именно в значении предсказание структуры. Правда, это делается в узких кругах, и в приличном обществе вслух такое не говорят. Так что можно считать, что пресс-релиз DeepMind просто содержал не к месту употребленный жаргонизм. Во-вторых, если прочитать этот релиз чуть дальше первого абзаца, его авторы как раз поясняют, что решали они именно задачу предсказания структуры, без всяких там процессов. Ну, это все демагогия и борьба за чистоту языка. Давайте перейдем к во-вторых и к размышлениям по делу.
А именно к размышлениям о слове «решение». Когда речь идет об использовании нейросетей для определения лиц или подсчета количества котиков на картинке, мы всегда можем понять, ошиблась нейросеть или нет. Просто потому что большинство людей способны по картинке определить, сколько на ней котиков и тот ли на ней человек, которого они перед собой видят. А вот когда мы используем нейросеть для решения задачи, ответа на которую не знаем, возникают вопросы. Как мы поймем, что нейросеть сработала верно и не сломалась? С какой долей уверенности мы можем использовать результаты работы этой нейросети в своей практической деятельности? Например, в проекте по разработке лекарственных препаратов, провал которого стоит несколько миллионов (иногда даже долларов)? Пока у нас нет ответа на этот вопрос, следовательно, говорить о решении, наверное, рановато.
Конечно, и в первой, и во второй версиях AlphaFold упоминается скор, призванный отличить хорошую работу алгоритма от плохой. Но вот данных по тому, насколько он соотносится с реальностью в открытом доступе, к сожалению, нет. Так что о практическом и рутинном применении речи, увы, пока не идет.
И тем не менее. Нельзя не отметить, что этот алгоритм показал работу, значительно превосходящую по результату все предыдущие попытки решить задачу предсказания структуры белка. И это означает, что потенциал метода, лежащего в основе этого алгоритма — огромный, и применять подобный метод можно не только к этой наукоемкой и сложной задаче. А значит, при должном развитии таких подходов нас ждет очень светлое и увлекательное будущее!
Заключение
В заключение хочется сказать следующее — AlphaFold2, безусловно, прорыв. Пока что есть некоторые моменты, ограничивающие его практическое применение, но, кажется, их можно преодолеть. Разумеется, для этого нужно задаться такой целью и уверенно к ней идти. И хочется верить, что DeepMind не бросит свое детище и будет и дальше его развивать и улучшать. Будет очень здорово, если это развитие будет проходить такими же семимильными шагами, как до этого.
Постзаключение
Надеемся, вас впечатлили существующие задачи, подходы к их решению и успехи этих подходов. На наш взгляд, самое красивое в таких событиях — это демонстрация того, как правильно примененные знания из разных областей могут дать невероятные результаты и помочь в решении сложнейших задач, стоящих перед человечеством.
AlphaFold появился на свет благодаря тому, что члены его команды обладали компетенциями в области биологии, физики, математики, алгоритмов глубокого обучения и оптимизации — то есть в области вычислительной биологии. Этому мало где хорошо учат в мире и пока что нигде на достаточном уровне не учат в России. Но в 2021 году в Высшая Школа Экономики совместно с компанией BIOCAD запускает магистерскую программу «Вычислительная биология и биоинформатика», в которой будут учить этим дисциплинам, столь необходимым для решения таких амбициозных задач.
В магистратуре ждут студентов с сильным физико-математическим бэкграундом, без химической и биологической подготовки. Преподаватели ВШЭ обеспечат лучшие в стране курсы по алгоритмам, программированию, анализу данных, а сотрудники индустрии расскажут спецглавы физики, молекулярной биологии, химии, а также специальные курсы по молекулярному моделированию, алгоритмам структурной биоинформатики, системной фармакологии и иным важным для области темам. И, что тоже важно, расскажут о настоящих индустриальных биологических задачах и научат использовать полученные знания и навыки для их решения.


