
Прежде чем перейти к статье, хочу вам представить, экономическую онлайн игру Brave Knights, в которой вы можете играть и зарабатывать. Регистируйтесь, играйте и зарабатывайте!
Привет, Хабр!
Меня зовут Елена Картышева, я выпускница программы «Разработка программного обеспечения» Университета ИТМО — одной из образовательных инициатив JetBrains. Я занимаюсь биоинформатикой и машинным обучением, и сегодня хочу рассказать о своей выпускной квалификационной работе. В рамках диплома я улучшила модель предсказания совместных побочных эффектов лекарств. Это поможет разрабатывать более эффективный дизайн клинических исследований и собирать информацию о побочных эффектах новых препаратов.

В этом году я окончила корпоративную магистерскую программу JetBrains в Университете ИТМО «Разработка программного обеспечения». Сейчас я работаю исследователем в лаборатории Machine Learning Applications and Deep Learning JetBrains Research.
Как я попала в магистратуру
В школе я хотела заниматься прикладной математикой и не особо любила программировать, поэтому поступила на направление «Прикладная математика и информатика» матмеха СПбГУ. На 4-м курсе пошла учиться в Институт Биоинформатики, заинтересовалась биологическими задачами и решила в дальнейшем развиваться в этом направлении. Там же я попала на стажировку в исследовательскую группу BioLabs в JetBrains Research, где занималась разработкой статистической модели для анализа данных ChIP-seq на Kotlin.
Мне нравилось работать со статистическими моделями, но из-за слабых навыков кодинга было сложно выполнять техническую часть работы. К счастью, мне повезло с научным руководителем, Алексеем Диевским, который много рассказывал про Kotlin и вообще JVM, и это оказалось неожиданно интересно. Я и раньше задумывалась, что для работы с прикладными задачами стоит научиться нормально программировать, но стажировка сыграла в этом решающую роль.
Так и получилось, что после окончания бакалавриата я решила поступать в корпоративную магистратуру JetBrains «Разработка программного обеспечения». Во-первых, мне ее посоветовал научный руководитель, а во-вторых, я знала о программе от друга, который учился на курс старше и рассказывал о серьезной нагрузке. Если честно, я думала, что не смогу поступить: про языки программирования я знала ужасно мало, да и с алгоритмами, которые являются важной частью собеседования, совсем не дружила. Но получилось как в меме: «Я по приколу подала заявку, меня по приколу взяли, и прикол вышел из-под контроля».
Пост получился довольно большим, поэтому вначале привожу краткий план своего рассказа:
Зачем нужно исследовать побочные эффекты лекарств?
Формулировка задачи
Существующие модели и их ограничения
Как я решила доработать модель
Способы векторного представления молекулы
Модель TriVec
Изменение структуры графа
Результаты
Заключение
Зачем нужно исследовать побочные эффекты лекарств?
Вам наверняка известно такое понятие, как побочные эффекты лекарств. Обычно различают индивидуальные побочные эффекты — это негативные последствия, вызванные приемом одного лекарства, и совместные — побочные эффекты, появляющиеся при приеме нескольких препаратов.
Совместные побочные эффекты возникают из-за нежелательного взаимодействия комбинаций препаратов. Они могут вызывать серьезные осложнения при лечении тяжелых пациентов [1]. Их сложно идентифицировать экспериментально: проверить все существующие пары препаратов попросту невозможно, поэтому в клинических испытаниях совместные побочные эффекты не тестируются, и информация о них появляется только когда лекарство начинают применять [2].
Формулировка задачи
Задачу определения побочных эффектов лекарств можно довольно удобно переформулировать как предсказание наличия или отсутствия ребра в графе знаний:

В лекарственном графе каждая вершина представляет собой то или иное лекарство. Если два препарата имеют совместный побочный эффект, то между соответствующими вершинами проводится ребро. В качестве дополнительной информации в граф также вводят вершины-белки и два новых типа взаимодействия: лекарство-белок и белок-белок. Это делают потому, что часто в основе фармакологического действия лекарства лежит взаимодействие препаратов с белками, соответственно они могут помочь в определении совместных побочных эффектов.
В итоге получается следующий граф: каждое ребро представляется тройкой (h, r, t), где h и t — вершины, которые соединяются ребром, а r — тип ребра: один из побочных эффектов или отображение связи лекарство-белок / белок-белок. Задача предсказания сводится к задаче бинарной классификации: необходимо определить, существует ли ребро (h, r, t) в графе.
Существующие модели и их ограничения
Через граф знаний задачу предсказания совместных побочных эффектов впервые сформулировали в 2018 году [3], и с тех пор было предложено достаточно много моделей. Одна из лучших — TriVec [4] достигает высоких значений метрик: AUROC (area under ROC curve) — 0.975, AUPRC (area under precision-recall curve) — 0.966 на датасете Decagon[5]. В этом датасете содержатся данные из баз побочных эффектов TWOSIDES [6], OFFSIDE [6] и SIDER [7], а также данные о белках, полученные из баз данных STITCH [8] и STRING [9].
Однако у существующих моделей есть ограничения: чем меньше побочных эффектов для лекарства мы знаем, тем хуже предсказания моделей. А для препаратов без известных побочных эффектов такие модели вообще не могут ничего предложить.
Результаты работы некоторых моделей на лекарствах без известных взаимодействий:
Модель | AUROC | AUPRC |
TriVec | 0.44 ± 0.016 | 0.535 ± 0.007 |
ComplEx | 0.352 ± 0.004 | 0.481 ± 0.004 |
Decagon | 0.303 ± 0.009 | 0.453 ± 0.004 |
Лекарств с малым количеством взаимодействий достаточно много — около 17% датасета. Также каждый год на фармацевтический рынок выходят новые препараты, для которых совместные побочные эффекты неизвестны вообще. Для таких случаев и нужны более совершенные модели, способные их предсказывать.
Один из сценариев использования подобных моделей — разработка схемы клинических испытаний. Когда производят новые препараты, проводят клинические тестирования в том числе и для выявления возможных побочных эффектов лекарства. Но исследований совместных побочных эффектов не делают, поскольку возможное количество испытаний слишком велико. Здесь и поможет модель: в дизайн эксперимента можно включить тестирование предсказаний, которые модель посчитает правдоподобными.
Как я решила доработать модель
Мы решили компенсировать недостаток информации добавлением в TriVec данных о химической структуре и схожести молекул. Я изучила разные подходы и остановилась на идее использовать векторные представления молекул для подсчета схожести веществ и внедрения ребер новых типов. Далее я подробно опишу, как это работает.
Способы векторного представления молекулы
Для того чтобы каким-либо образом считать схожесть молекул, нужно каждое лекарство представить в виде числового вектора, который будет отражать химические свойства препарата. Сделать это можно множеством способов, но есть проблема. Каждый из способов будет учитывать только часть свойств, например, структуру молекулы, ее биофизические свойства или одиночные побочные эффекты.
Различаются и способы получения представлений. Какие-то просто являются набором измерений различных свойств (например, молекулярные дескрипторы), какие-то основаны на простых и достаточно понятных алгоритмах (например, различные алгоритмы получения молекулярных отпечатков пальцев), а какие-то получены с помощью моделей машинного обучения (например, модели HVAE [10] и ChemBERTa [11]), и неизвестно, как именно в них представлена химическая информация.
Мне было довольно сложно выбрать подходящее представление из такого зоопарка. Я биоинформатик, поэтому понимаю основы биологии и работы разных биологических моделей, но химия для меня — темный лес. Где-то я консультировалась со знакомыми и научными руководителями, где-то находила хорошие статьи с пояснениями для чайников, но точно могу сказать, что эта часть работы была для меня одной из самых сложных.
В итоге я остановилась на трех типах векторного представления молекул, которые пробовала для дальнейшего использования:
1. Молекулярные отпечатки пальцев по Моргану [12]. Это один из самых распространенных типов векторного представления. Молекула отображается как битовый вектор, где каждый бит кодирует наличие или отсутствие определенной подструктуры.

2. Молекулярные дескрипторы [13]. Вектор представляет собой набор значений различных молекулярных свойств, например, атомной массы и заряда. Это представление широко используются в разных задачах предсказания молекулярных свойств, поэтому его применение может помочь и в текущей задаче.
3. Векторное представление из модели HVAE [14] — Hyperbolic Variational Autoencoder.
Быстро напомню идею модели автоэнкодера: модель пытается научиться отображать входные данные в латентное пространство и обратно.

Одна из вариаций такой модели — это как раз HVAE, использующая в качестве скрытого пространства гиперболическое. На вход модели подается структура молекулы в виде SMILES-строки [15].

Кроме информации о структуре, модель во время обучения использует информацию о положении лекарства в иерархии лекарств ATC [16]. Эта иерархия учитывает терапевтическое и фармакологическое действие, а также химическую группу препарата. Модель пытается представить в латентном пространстве лекарство таким образом, чтобы схожие по структуре и действию на организм препараты были как можно ближе друг к другу.

На схожих задачах, таких как предсказание взаимодействия лекарства и белка, использование этого представления дало хорошие результаты, поэтому я решила попробовать его и в дипломной работе.
Модель TriVec
Модель TriVec в своей основе использует принцип матричного разложения.
Модели такого типа устроены по одному принципу. Каждой вершине i сопоставляется векторное представление (эмбеддинг) ei ∈ Rd, каждому ребру сопоставляется эмбеддинг wi ∈ Rl. Обучающие данные состоят из троек (h, r, t), где h — вершина из которой исходит ребро, t — вершина в которую входит ребро, а r — тип ребра. У каждой модели есть функция оценки f(h, r, t), которая показывает правдоподобность тройки: чем выше значение функции, чем более правдоподобна тройка. Функция оценки использует эмбеддинги вершин eh, et и ребра wr для подсчета значения. Обучение происходит путем минимизации функции потерь L которая вычисляется по значениям функции оценки на истинных тройках (существующих в графе) и ложных (негативных).
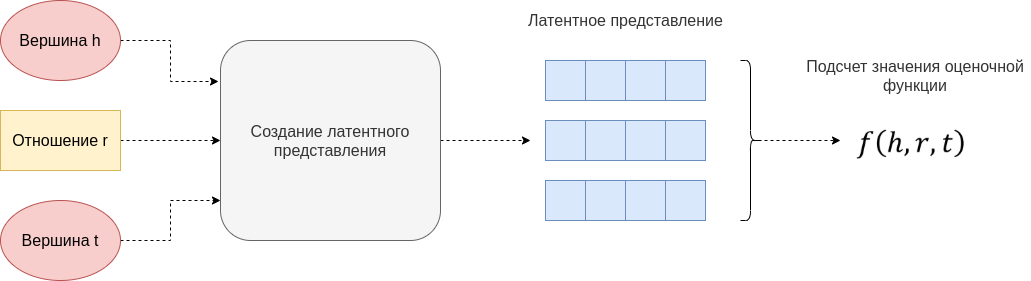
В модели TriVec каждая вершина i и ребро j имеют по три эмбеддинга заданного размера K (обозначаются как (e1i , e2i, e3i) и (w1i , w2i, w3i)). То есть эмбеддинги вершин и ребер имеют размер K3. Эмбеддинги инициализируются случайно.
Модель обучается итеративно, на каждом шаге (эпохе) обновляя эмбеддинги таким образом, чтобы минимизировать функцию потерь модели, которая имеет следующий вид:


где E — множество всех сущностей.
Функция оценки TriVec выглядит следующим образом:

где <...> — скалярное произведение.
Изменение структуры графа
После этого мне нужно было выбрать способы, как добавить информацию в модель. Здесь мне здорово помогла мой научный руководитель Нина Лукашина. Мы обсуждали, пробовали, меняли и пробовали снова. Это было время бесконечных вычислительных экспериментов, многие из которых проваливались.
В итоге мы остановились на трёх способах улучшения модели.
1. Изменение случайной инициализации эмбеддингов вершин и ребер на химическое представление.
Этот способ был выбран, как самое простое и распространенное улучшение. В качестве инициализирующих векторов использовалось одно из трех векторных представлений, описанных выше (тип представления является параметром).
2. Добавление в граф нового типа ребра с весом.
Введение такого ребра позволит распространять информацию между похожими лекарствами. Идея состоит в том, чтобы вес зависел от химической схожести молекул следующим образом:
чем более схожи два лекарства, тем вес больше;
чем больше степень вершины, тем вес меньше.
Обратная пропорциональность степени вершины и веса обусловлена следующим: на лекарствах с большим количеством связей модель и так показывает достаточно хорошие результаты, поэтому улучшение обучения необходимо только для вершин со сравнительно небольшими степенями.
Для подсчета схожести мы использовали векторные представления молекул. Поскольку часто эти представления рассчитаны как раз на то, чтобы считать схожесть молекул, им обычно соответствует некая функция схожести. Для молекулярных отпечатков пальцев по Моргану это коэффициент Танимото, а для векторов, полученных из модели HVAE, — близость в гиперболическом пространстве.
Определим функцию потерь для нового типа ребер следующим образом:

где rweighted — новый тип ребра.
Для подсчета веса в случае молекулярных отпечатков пальцев по Моргану и векторов, полученных из модели HVAE:


а similarity(h,t) — функция схожести, соответствующая выбранному типу векторного представления.
Поскольку для молекулярных дескрипторов нет естественной функции схожести, её пришлось подбирать. В итоге я остановилась на следующем способе:

где window_size(h,t) = lower_bound+inverse_deg(h)*inverse_deg(t)*(upper_bound-lower_bound), lower_bound=0.5, upper_bound=20 (так же подбирались исходя из данных).
Подводя итог, в этом улучшении параметром также является тип векторного представления. C молекулярными отпечатками пальцев по Моргану и векторами, полученными из модели HVAE, использовалась функция weight1для подсчета весов, для молекулярных дескрипторов —weight2, поскольку для них нет естественной функции схожести.
3. Приближение эмбеддингов лекарств с малым количеством взаимодействий к топ-N лекарств, похожим по одиночным побочным эффектам, с помощью MSE loss.
Несмотря на то, что для многих лекарств выполняется предположение, что похожие по структуре и свойствам вещества имеют похожее действие, для части препаратов это не так. Одиночные побочные эффекты гораздо сильнее связаны с совместными побочными эффектами, поэтому у меня возникла идея использовать одиночные побочные эффекты для распространения информации.
Алгоритм такой: для каждого лекарства берется топ-???? самых близких веществ по одиночным побочным эффектам. На каждой итерации для всех слабых лекарств идет приближение к этим топ-???? с помощью метода наименьших квадратов. Таким образом, эмбеддинги лекарств с недостаточным количеством связей или совсем без них будут приближаться к эмбеддингам самых похожих на них лекарств по одиночным побочным эффектам.

Результаты
Чтобы тестировать дальнейшие улучшения, я сформировала из исходного датасета два новых разбиения на обучающую, валидационную и тестовую выборки. В первом из них в тесте содержатся только примеры о лекарствах с малым количеством взаимодействий, а во втором — только о лекарствах без известных взаимодействий.
Для каждого типа улучшения я выбирала векторное представление и значение параметра N, дающее наилучшие результаты. Эти результаты представлены в таблицах ниже. Каждая модель запускалась десять раз, в таблице представлено среднее значение и отклонение результатов запусков.
В качестве метрик использовались AUROC (area under ROC curve) и AUPRC (area under precision-recall curve).
Результаты получились достаточно хорошими. Поскольку для лекарств с малым количеством побочных эффектов эмбеддинги вообще не обновлялись в процессе обучения, предсказания исходной модели стали хуже, чем у случайного классификатора (0.44 AUROC). Улучшения позволили нам достичь довольно хорошей предсказательной способности (0.77 AUROC). На лекарствах без известных взаимодействий предсказание исходной модели упали до 0.85 AUROC, внедрение улучшений позволило поднять предсказания до 0.895 AUROC.


Заключение
Несмотря на то, что моя модель позволила довольно сильно улучшить результаты на лекарствах с малым количеством связей и без известных связей, есть довольно много идей для дальнейшего развития этой темы.
К сожалению, мне не хватило времени на подробное исследование результатов. Хочется посмотреть, какие свойства отражают в себе полученные эмбеддинги лекарств, проверить, у какой доли новых предсказаний найдется подтверждение в литературных источниках, собрать новую тестовую выборку из лекарств без известных взаимодействий и протестировать модель на ней. Кроме этого, у модели есть довольно серьезный недостаток: она не является индуктивной, то есть при добавлении новой вершины или типа ребра нужно будет полностью ее переобучать.
Исследованием этой задачи мы планируем и дальше заниматься в лаборатории, тема взаимодействия лекарств, белков и других компонентов очень интересная и обширная. Задач для изучения довольно много: сделать модель более детальной (добавить различные дозировки лекарств, частоту побочных эффектов), расширить модель предсказания ребра с маленького лекарственного графа на более обширные биологические графы, и многие другие.
Писать диплом было сложно. Он отнимал много времени, очень много времени! В третьем семестре работа над дипломом занимала около 20 часов в неделю, в последнем на него уходил почти каждый день. Тем не менее, мне очень понравилось заниматься этой темой: приходилось самой искать разные статьи, изучать, предлагать идеи развития проекта. Работа над дипломом дала мне много навыков, важных для работы исследователем.
А в конце хочу выразить благодарность своим научным руководителям Нине Лукашиной и Алексею Александровичу Шпильману. Мы много общались с ними по ходу работы, обсуждали идеи, готовили презентацию и текст диплома. Я считаю, что мне с ними очень повезло!
До 9 августа продолжается прием документов на корпоративную магистерскую программу JetBrains «Разработка программного обеспечения». В этом году набор ведется на 30 бюджетных и 5 платных мест. Подробнее о программе можно узнать на сайте или в Telegram-чате для абитуриентов.
Источники:
[1] Trends in Prescription Drug Use Among Adults in the United States From 1999-2012 / Elizabeth Kantor, Colin Rehm, Jennifer Haas et al. // JAMA. –– 2015. –– 11. –– Vol. 314. –– P. 1818–1830.
[2] Bansal, M. et al. (2014). A community computational challenge to predict the activity of pairs of compounds. Nature Biotechnology, 32(12), 1213–1222.
[3] Zitnik Marinka, Agrawal Monica, Leskovec Jure. Modeling polypharmacy side effects with graph convolutional networks // Bioinformatics. — 2018. — Vol. 34, no. 13. — P. 457–466.
[4] Novacek Vit, Mohamed Sameh. Predicting Polypharmacy Side-effects Using Knowledge Graph Embeddings // AMIA Joint Summits on Translational Science proceedings. AMIA Joint Summits on Translational Science. — 2020. — 05. — Vol. 2020. — P. 449–458.
[5] Zitnik,M. et al. BioSNAP datasets: Stanford biomedical network dataset collection. — 2018.
[6] Tatonetti, N. P. et al. (2012). Data-driven prediction of drug effects and interactions. Science Translational Medicine, 4(125), 12531.
[7] Kuhn, M. et al. (2015). The SIDER database of drugs and side effects. Nucleic Acids Res., 44(D1), D1075–D1079
[8] Szklarczyk, D. et al. (2015). STITCH 5: augmenting protein–chemical interaction networks with tissue and affinity data. Nucleic Acids Res., 44(D1), D380–D384.
[9] Szklarczyk, D. et al. (2017). The STRING database in 2017: quality-controlled protein–protein association networks, made broadly accessible. Nucleic Acids Res., 45(D1), D362–D368.
[10] A wrapped normal distribution on hyperbolic space for gradient-based learning / Yoshihiro Nagano, Shoichiro Yamaguchi, Yasuhiro Fujita, Masanori Koyama // International Conference on Machine Learning / PMLR. –– 2019. –– P. 4693–4702.
[11] Chithrananda Seyone, Grand Gabriel, Ramsundar Bharath. ChemBERTa: Large-Scale Self-Supervised Pretraining for Molecular Property Prediction // CoRR. –– 2020. –– Vol. abs/2010.09885. –– 2010.09885.
[12] A Practical Introduction to the Use of Molecular Fingerprints in Drug Discovery
[13] Aguayo-Ortiz Rodrigo, Fernandez-de Gortari Eli. Chapter 2 - Overview of Computer-Aided Drug Design for Epigenetic Targets // Epi-Informatics / Ed. by Jose L. Medina-Franco. –– Boston: Academic Press, 2016. –– P. 21–52. –– Access mode: https://www.sciencedirect.com/science/article/pii/B9780128028087000022.
[14] Yu Ke, Visweswaran Shyam, Batmanghelich Kayhan. Semi-supervised hierarchical drug embedding in hyperbolic space // Journal of Chemical Information and Modeling. –– 2020.
[15] Weininger David. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules // Journal of Chemical Information and Modeling. –– 1988. –– Feb. –– Vol. 28, no. 1. –– P. 31–36.
[16] "Purpose of the ATC/DDD system". WHO Collaborating Centre for Drug Statistics Methodology. Retrieved 6 July 2021.
Интересные статьи
Интересные статьи



