
Прежде чем перейти к статье, хочу вам представить, экономическую онлайн игру Brave Knights, в которой вы можете играть и зарабатывать. Регистируйтесь, играйте и зарабатывайте!

Биология человека – невероятно сложная наука. Даже учитывая, что с каждым годом мы открываем всё больше секретов человеческого тела, получаемые нами ответы порождают всё большее количество вопросов. Завершение проекта "Геном человека" придало многим учёным уверенность в том, что с помощью геномики человечество сможет решить важные биологические проблемы. Однако, чем больше биологических тайн мы раскрываем, тем более отчётливо понимаем, что на использование генома организма влияют другие факторы. Соответственно, для решения задач в этих взаимосвязанных областях, в том числе транскриптомике (изучение мРНК) и протеомике (изучение белков) были созданы новые направления научных исследований, в которых начали использовать Python.
Сегодня, специально к старту новых потоков на курсы Python для анализа данных, Fullstack-разработчик на Python и Python для веб-разработки в этой статье расскажем вам про еще одно применение этого прекрасного языка — в научных исследованиях.
Проект Biopython – набор инструментов Python для вычислительной биологии и биоинформатики – используется для визуализации и анализа последовательностей ДНК и РНК. С помощью набора инструментов Biopython также можно выполнять анализ белковых структур! Познакомимся с ним более подробно.
Банк белковых структур (PDB) – единая база данных для изучения и загрузки последовательностей белка. Для работы с PDB был создан специальный файловый формат, естественно, получивший название .pdb. Но по мере того как учёным приходилось анализировать более крупные и сложные белковые структуры, были разработаны другие форматы - CIF и mmCIF. Файл кристаллографической информации CIF (Crystallographic Information File) был разработан для архивирования данных кристаллографического исследования малых молекул. В рамках таких исследований изучается расположение атомов в кристаллических твердых телах. Со временем формат CIF стал использоваться для анализа более крупных молекул (макромолекул, отсюда обозначение mm), получил название mmCIF и в итоге заместил формат PDB. [1]
Визуализация данных с помощью формата PDB
Несмотря на то что в настоящее время общепринятым стандартом является формат mmCIF, многие системы по-прежнему поддерживают файлы старого формата PDB.
Рассмотрим Фитогемагглютинин-L – лектин, содержащийся в некоторых бобовых, например в стручковой фасоли.
Импортируйте необходимые пакеты:
from Bio.PDB import *
import nglview as nv
import ipywidgetsТеперь создадим экземпляр PDBParser Biopython и для создания интерактивной визуализации воспользуемся библиотекой nglview. Мы можем панорамировать, масштабировать и вращать молекулу и даже выводить на экран определённую информацию об атомах.
pdb_parser = PDBParser()
structure = pdb_parser.get_structure("PHA-L", "Data/1FAT.pdb")
view = nv.show_biopython(structure)
Визуализация данных с помощью формата CIF
Процедура для файлов CIF практически такая же, за тем исключением, что нужно использовать экземпляр MMCIF Parser! Здесь мы визуализируем более крупную белковую структуру 6EBK, или канал paddle chimera Kv1.2-2.1 в липидных нанодисках (даже произносить трудно…).
cif_parser = MMCIFParser()
structure = cif_parser.get_structure("6EBK", "fa/6ebk.cif")
view = nv.show_biopython(structure)Доступ к информации о белковой структуре
Заголовок
Самый быстрый способ доступа к информации о белковой структуре – через заголовок, словарь метаданных, доступный как в формате PDB, так и в формате CIF.
mmcif_dict = MMCIFDict.MMCIFDict("fa/1fat.cif")
len(mmcif_dict) # 689В результате создаётся большой словарь информации о белковой структуре, в том числе цитата, определяющая последовательность белка, информацию о структуре, расположениях и углах атомов, а также химический состав. Как видите, словарь состоит из 689 позиций.
Последовательности остатков
Одна из самых важных частей анализируемой информации – это последовательность остатков белка или полипептида (аминокислот). Поскольку белки могут состоять из нескольких полипептидов, для понимания исследуемого уровня организации мы используем структурный подход. От общей структуры – до отдельных атомов.
Объект Structure в нашем файле построен в архитектуре SMCRA (СМЦОА) в соответствии со схемой “родитель – дочерний элемент”:
Структура состоит из моделей.
Модель состоит из цепочек.
Цепочка состоит из остатков (аминокислот).
Остаток состоит из Атомов.
Для получения последовательностей остатков белка существует множество способов разбора метаданных структуры. Рассмотрим три варианта:
# .get_residues() method in a loop
for model in structure:
for residue in model.get_residues():
print(residue)
# .get_residues() method as generator object
residues = structure.get_residues() # returns a generator object
[item for item in residues]
# .unfold_entities - keyword for each level of the SMCRA structure
Selection.unfold_entities(structure, "R") # R is for residuesСоздание полипептидов
Получение упомянутой выше последовательности остатков возвращает последовательность для всей белковой структуры, однако белки часто состоят из нескольких полипептидов меньшего размера, которые, возможно, стоит проанализировать отдельно. Набор инструментов Biopython позволяет сделать это с помощью построителей полипептидов, генерирующих отдельные полипептиды.
polypeptide_builder = CaPPBuilder()
counter = 1
for polypeptide in polypeptide_builder.build_peptides(structure):
seq = polypeptide.get_sequence()
print(f"Sequence: {counter}, Length: {len(seq)}")
print(seq)
counter += 1
# Sequence: 1, Length: 36
# SNDIYFNFQRFNETNLILQRDASVSSSGQLRLTNLN
# Sequence: 2, Length: 196
# NGEPRVGSLGRAFYSAPIQIWDNTTGTVASFATSFT...ASKLS
# Sequence: 3, Length: 233
# SNDIYFNFQRFNETNLILQRDASVSSSGQLRLTNLN...ASKLS
# Sequence: 4, Length: 36
# SNDIYFNFQRFNETNLILQRDASVSSSGQLRLTNLN
# Sequence: 5, Length: 196
# NGEPRVGSLGRAFYSAPIQIWDNTTGTVASFATSFT...ASKLS
# Sequence: 6, Length: 35
# SNDIYFNFQRFNETNLILQRDASVSSSGQLRLTNL
# Sequence: 7, Length: 196
# NGEPRVGSLGRAFYSAPIQIWDNTTGTVASFATSFT...ASKLSАнализ последовательностей остатков
Итак, теперь у нас есть последовательности остатков для этих 7 цепочек, и такие последовательности можно проанализировать множеством методов.
from Bio.SeqUtils.ProtParam import ProteinAnalysisЕдинственное предостережение: вызов процедуры .get_sequences() возвращает объект Biopython Seq(), см. предыдущую запись в моем блоге, где приведено более подробное описание объектов Seq() и их функциональности – ProteinAnalysis требует строкового значения.
analyzed_seq = ProteinAnalysis(str(seq))Теперь мы готовы к запуску следующих методов, которые позволят понять нашу последовательность!
Молекулярный вес
Мы можем рассчитать молекулярный вес полипептида.
analyzed_seq.molecular_weight()
# 4176.51669GRAVY
Белок GRAVY возвращает значение GRAVY (общее среднее значение гидропатии) для введённых белковых последовательностей. Значение GRAVY вычисляется путём сложения значения гидропатии для каждого остатка и деления на длину последовательности (Kyte и Doolittle; 1982). [2]
Более высокое значение указывает на большую гидрофобность. Меньшее – на большую гидрофильность. Позже мы обсудим, как генерировать остаток по гидрофобности остатков.
analyzed_seq.gravy()
# -0.5611Подсчёт количества аминокислот
Количество аминокислот каждого типа можно легко подсчитать.
analyzed_seq.count_amino_acids()
# {'A': 1,
'C': 0,
'D': 2,
'E': 1,
'F': 3,
'G': 1,
'H': 0,
'I': 2,
'K': 0,
'L': 5,
'M': 0,
'N': 6,
'P': 0,
'Q': 3,
'R': 3,
'S': 5,
'T': 2,
'V': 1,
'W': 0,
'Y': 1}Процент аминокислот
А также процентную долю каждой аминокислоты в последовательности!вставка с кодом
Вторичная структура
Очень полезный метод – .secondary_structure_fraction() – возвращает долю аминокислот, которые могут быть обнаружены в трёх классических вторичных структурах. Это бета-складчатые структуры, альфа-спирали и петли (на которых остатки меняют направление).
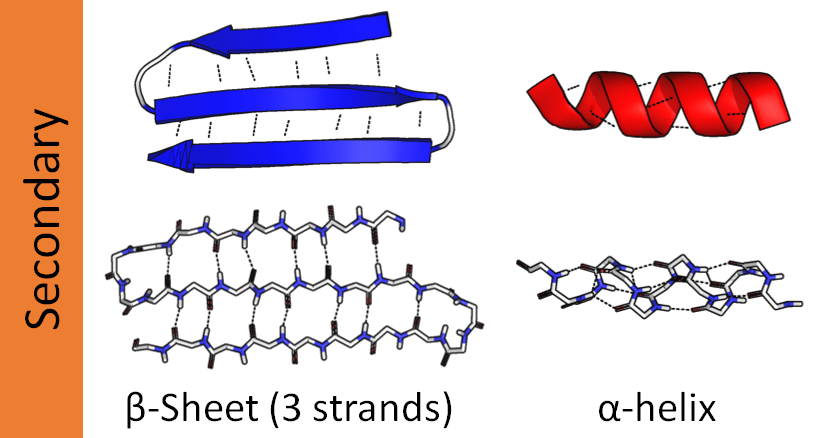
analyzed_seq.secondary_structure_fraction() # helix, turn, sheet
# (0.3333333333333333, 0.3333333333333333, 0.19444444444444445)Протеиновые весы
Протеиновые весы – это способ измерения определённых атрибутов остатков по длине последовательности пептидов с помощью "скользящего" окна. Весы градуируются значениями для каждой аминокислоты. Каждое значение базируется на различных физических и химических свойствах, таких как гидрофобность, тенденции вторичной структуры и доступность поверхности. В отличие от некоторых единиц измерения на уровне цепочки, таких как общее поведение молекул, весы позволяют более детально понять, как будут вести себя более мелкие участки последовательности.
from Bio.SeqUtils.ProtParam import ProtParamDataВот некоторые распространённые весы:
kd → индекс гидрофобности Kyte & Doolittle [оригинал статьи];
Flex → нормализованные средние параметры гибкости (B-значения) [оригинал статьи];
hw → индекс гидрофобности Hopp & Wood [оригинал статьи];
em → поверхностная дробная вероятность Emini (доступность поверхности) [оригинал книги].
Документацию по некоторым распространённым весам можно найти здесь.
В качестве примера рассмотрим индекс гидрофобности (kd). Представлены весы, в которых каждый остаток имеет связанное значение, представляющее уровень его гидрофобности.
kd = {"A": 1.8, "R": -4.5, "N": -3.5, "D": -3.5, "C": 2.5,
"Q": -3.5, "E": -3.5, "G": -0.4, "H": -3.2, "I": 4.5,
"L": 3.8, "K": -3.9, "M": 1.9, "F": 2.8, "P": -1.6,
"S": -0.8, "T": -0.7, "W": -0.9, "Y": -1.3, "V": 4.2}Положительные значения означают гидрофобность. Изолейцин (I) и валин (V) наиболее гидрофобны, а аргинин (R) и лизин (K) наиболее гидрофильны. Гидрофобные остатки, как правило, находятся внутри полипептида, а гидрофильные остатки – за его пределами, поэтому эти весы также дают представление о том, как может складываться такой полипептид.
Чтобы провести анализ на основе белковых весов, необходимо задать размер окна, в котором будет вычисляться среднее значение. Используя ключевое слово "edge", можно также измерять важность соседних остатков, в основном определяя их важность по отношению к среднему для окна значению.
analysed_seq.protein_scale(window=7, param_dict=ProtParamData.kd)
# [-0.7571428571428572,
-0.2428571428571429,
-0.24285714285714288,
-0.38571428571428573,
-0.6285714285714287,
-0.942857142857143,
-1.842857142857143,
-1.442857142857143,
-2.3428571428571425,
-1.3000000000000003,
-0.01428571428571433,
0.1285714285714285,
0.1285714285714285,
-0.014285714285714235,
-0.4142857142857143,
0.3428571428571428,
-0.31428571428571417,
-0.35714285714285715,
-1.014285714285714,
-0.6285714285714284,
-0.10000000000000002,
0.3428571428571429,
-0.4142857142857142,
0.24285714285714285,
-1.0,
-0.34285714285714286,
-0.32857142857142857,
-0.7142857142857143,
-0.1142857142857144,
-0.11428571428571435]Попробуем вместе
Давайте объединим все наши методы и создадим скрипт, который выполнит итерацию по каждой цепочке нашей структуры и запустит какой-нибудь стандартный анализ. Создадим пустой контейнер, заполним его словарём ключевой информации для каждой последовательности. После создания такой вложенной структуры мы сможем получать срезы, как и в любом контейнере на Python, отдельных записей.
# Create empty list for chains
all_seqs = []
counter = 1
# For each polypeptide in the structure, run protein analysis methods and store in dict
for pp in ppb.build_peptides(structure):
seq_info = {} # create an empty dict
seq = pp.get_sequence() # get the sequence like above
analyzed_seq = ProteinAnalysis(str(seq)) # needs to be a str
# Specify dict keys and values
seq_info['Sequence Number'] = counter # set sequence id
seq_info['Sequence'] = seq # store BioPython Seq() object
seq_info['Sequence Length'] = len(seq) # length of seq
seq_info['Molecular Weight'] = analyzed_seq.molecular_weight()
seq_info['GRAVY'] = analyzed_seq.gravy() # hydrophobicity
seq_info['AA Count'] = analyzed_seq.count_amino_acids()
seq_info['AA Percent'] = analyzed_seq.get_amino_acids_percent()
# tuple of (helix, turn, sheet)
seq_info['Secondary Structure'] = \
analyzed_seq.secondary_structure_fraction()
# Update all_seqs list and increase counter
all_seqs.append(seq_info)
counter += 1Выбор первой последовательности возвращает словарь с нашими анализами и значениями!
all_seqs[0] # select the first sequence
# {'Sequence Number': 1,
'Sequence': Seq('SNDIYFNFQRFNETNLILQRDASVSSSGQLRLTNLN'),
'Sequence Length': 36,
'Molecular Weight': 4176.52,
'GRAVY': -0.5611,
'Amino Acid Count': {'A': 1,
'C': 0,
'D': 2,
'E': 1,
'F': 3,
'G': 1,
'H': 0,
'I': 2,
'K': 0,
'L': 5,
'M': 0,
'N': 6,
'P': 0,
'Q': 3,
'R': 3,
'S': 5,
'T': 2,
'V': 1,
'W': 0,
'Y': 1},
'Amino Acid Percent': {'A': 0.027777777777777776,
'C': 0.0,
'D': 0.05555555555555555,
'E': 0.027777777777777776,
'F': 0.08333333333333333,
'G': 0.027777777777777776,
'H': 0.0,
'I': 0.05555555555555555,
'K': 0.0,
'L': 0.1388888888888889,
'M': 0.0,
'N': 0.16666666666666666,
'P': 0.0,
'Q': 0.08333333333333333,
'R': 0.08333333333333333,
'S': 0.1388888888888889,
'T': 0.05555555555555555,
'V': 0.027777777777777776,
'W': 0.0,
'Y': 0.027777777777777776},
'Secondary Structure': (0.3333333333333333,
0.3333333333333333,
0.19444444444444445)}Можно легко выбирать конкретные значения.
all_seqs[0]['Sequence']
# Seq('SNDIYFNFQRFNETNLILQRDASVSSSGQLRLTNLN')
all_seqs[0]['Molecular Weight']
# 4176.52Заключение
Набор инструментов Biopython не только облегчает работу с последовательностями ДНК, но его также можно использовать для целей протеомики для визуализации и анализа белков. Biopython реализует мощные и гибкие методы стандартного анализа белков, результаты которых могут быть использованы для разработки индивидуальных процессов в соответствии с вашими конкретными потребностями. Со временем мне, несомненно, станут известны новые впечатляющие функциональные возможности Biopython, и я опубликую множество статей на эту интересную тему.
Как и всегда, все коды и зависимости, описанные в данной статье, можно найти в этом репозитории, который я продолжу обновлять по мере изучения набора инструментов Biopython. Надеюсь, что данное руководство поможет вам запускать собственные проекты по биоинформатике с помощью Biopython!
А если ваши потребности далеки от биоинформатики — не беда, ведь Python весьма распространен в других направлениях и является просто мастхэвом например для дата-сайентистов и аналитиков. Для освоения Python у нас есть целых три курса, Python для анализа данных, Fullstack-разработчик на Python и Python для веб-разработки, каждый из которых имеет свою специфику, можно выбрать какой больше отвечает вашим целям. А промокод HABR даст скидку 50%.

Узнайте, как прокачаться и в других специальностях или освоить их с нуля:
Профессия Data Scientist
Профессия Data Analyst
Курс по Data Engineering
Другие профессии и курсы
ПРОФЕССИИ
Профессия Java-разработчик
Профессия QA-инженер на JAVA
Профессия Frontend-разработчик
Профессия Этичный хакер
Профессия C++ разработчик
Профессия Разработчик игр на Unity
Профессия Веб-разработчик
Профессия iOS-разработчик с нуля
Профессия Android-разработчик с нуля
КУРСЫ
Курс по Machine Learning
Математика и Machine Learning для Data Science
Математика для Data Science
Курс "Алгоритмы и структуры данных"
Курс по аналитике данных
Курс по DevOps



