
Прежде чем перейти к статье, хочу вам представить, экономическую онлайн игру Brave Knights, в которой вы можете играть и зарабатывать. Регистируйтесь, играйте и зарабатывайте!

Про биоинформатику слышали многие. Кто-то знает больше, кто-то меньше. Мы постарались раскрыть вопрос этой, относительно новой, науки. Так сказать, дать общие представления читателю об основных вехах развития, методах и проблемах: решённых и существующих на нынешнее время.
Автор сообщества Фанерозой, генетик, ботаник Степан Иванов.

С развитием информационных технологий появилось множество удобств самого разного характера. Не обошли стороной эти удобства и научные исследования. Совершенно неудивительно, что и в биологии появился и нашёл применение целый огромный раздел науки — биоинформатика. Хотя следует отметить, что это и произошло совсем недавно. Так, датой начала использования биоинформатики в биологии можно назвать 1970 год. Тогда Полина Хогевег (нидерландский биолог) и Бен Хеспер ввели термин «биоинформатика», определив его как изучение информационных процессов в биотических системах.
С этого момента биоинформатика развивалась стремительно и неумолимо. В 1970 году Нидельманом-Вуншем была предложена система сравнения аминокислотных и нуклеотидных последовательностей. Эти алгоритмы быстро находили применение в огромном количестве исследований и стали фундаментом для новых открытий. Одним из наиболее важных прорывов стали алгоритмы и программы, позволяющие предсказывать вторичные структуры белков, а на их основе — предсказывать функции отдельных доменов и белков в целом (мы писали об этом здесь).

Вообще, стандартным решением многих биологических вопросов является сравнение полученных данных с эталоном, и информационные технологии, позволяющие перенести эти расчёты на компьютеры. Исследования в этой области знания приносят очень прорывные и технологические плоды.
Визуализированный результат работы алгоритмов выравнивания аминокислотных последовательностей:
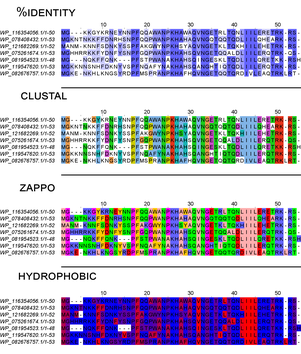
Вместе с развитием применения информационных технологий развивались и сами методы получения этих данных. Происходило их накопление. Например, на новый уровень выходили методы секвенирования, получали целые геномы модельных организмов, а их расшифровка и аннотирование во многом ложилось на вычислительную технику. Не углубляясь в историю развития методов NGS (next generation sequencing) отметим лишь, что сейчас есть возможность получать до 2 миллионов пар нуклеотидов за одно прочтение по весьма низкой себестоимости (в случае секвенирования методом MinION — стоимость одного миллиона пар нуклеотидов колеблется в районе 1$).

Чем больше становилось референсных данных, тем больше была точность работы программ. Конечно, развивались и алгоритмы, обрабатывающие эти данные. И постепенно интерес в биоинформатике сместился с изучения отдельных частиц к исследованию взаимодействий разных генов. Отсюда даже выделился новый раздел биоинформатики — системная биология (объект — как система связанных процессов и компонентов взаимодействующих друг с другом).

Новые технологии позволили получать все больше данных, в свою очередь, эти массивы охватить человеческим сознанием уже было невозможно, а многие расчёты на компьютерах до сих пор занимают дни и недели (например, сборки больших геномов).
Чтобы не быть голословным, приведём несколько примеров исследований с использованием методов биоинформатики:
- Предсказание трёхмерной структуры белкаТак, команда DeepMind буквально год назад решила вопрос по предсказанию трёхмерной структуры белка по его аминокислотной последовательности. Хотя сама проблема считалась буквально фундаментальной и сформулированной ещё в 1970-х.Сама идея о том, что аминокислотная последовательность белка несёт в себе информацию о его функциях была высказана нобелевским лауреатом по химии Кристианом Анфинсном. С тех пор это стало вызовом для компьютерных технологий, поскольку альтернативой для выяснения трёхмерной структуры (а значит, и функции белка) были лишь долгие эксперименты, тянувшиеся порой десятки лет для выяснения функции лишь одного белка. В 1969 году проблему обозначил Сайрус Левинталь, оценивший количество конформаций для типичного белка как 10 в 300 степени вариантов. Разработанный программистами ИИ AlphaFold позволяет весьма быстро (2–3 дня расчётов для типичного белка на не самом мощном компьютере) решать вопрос третичной конформации белка, а значит и его функции. Конечно, и здесь остаются проблемы, одна из основных — ИИ обучался на тех белках, для которых третичной структуры уже известны и верификация расчётов для отличных белков остаётся под вопросом. Но даже так, это однозначно прорыв в фундаментальной, структурной биологии. И, однозначно, найдёт своё применение в разработке многих лекарств. Больше можно почитать про ИИ на этом сайте .
- Сборка геномовНаверняка многие слышали про NGS — секвенирование нового поколения. Методы NGS позволяют получать огромные нуклеотидные последовательности. При этом для того или иного участка по итогу секвенирования может существовать много тысяч прочтений и выравнивание этих фрагментов относительно друг друга сделать человеческими силами просто невозможно. Однако же существует ряд программ, в которых любой исследователь может получить консенсусные последовательности, просто внеся нужные параметры и переложив все вычисления на компьютер.
- Биоинформатические методы в традиционных исследованиях
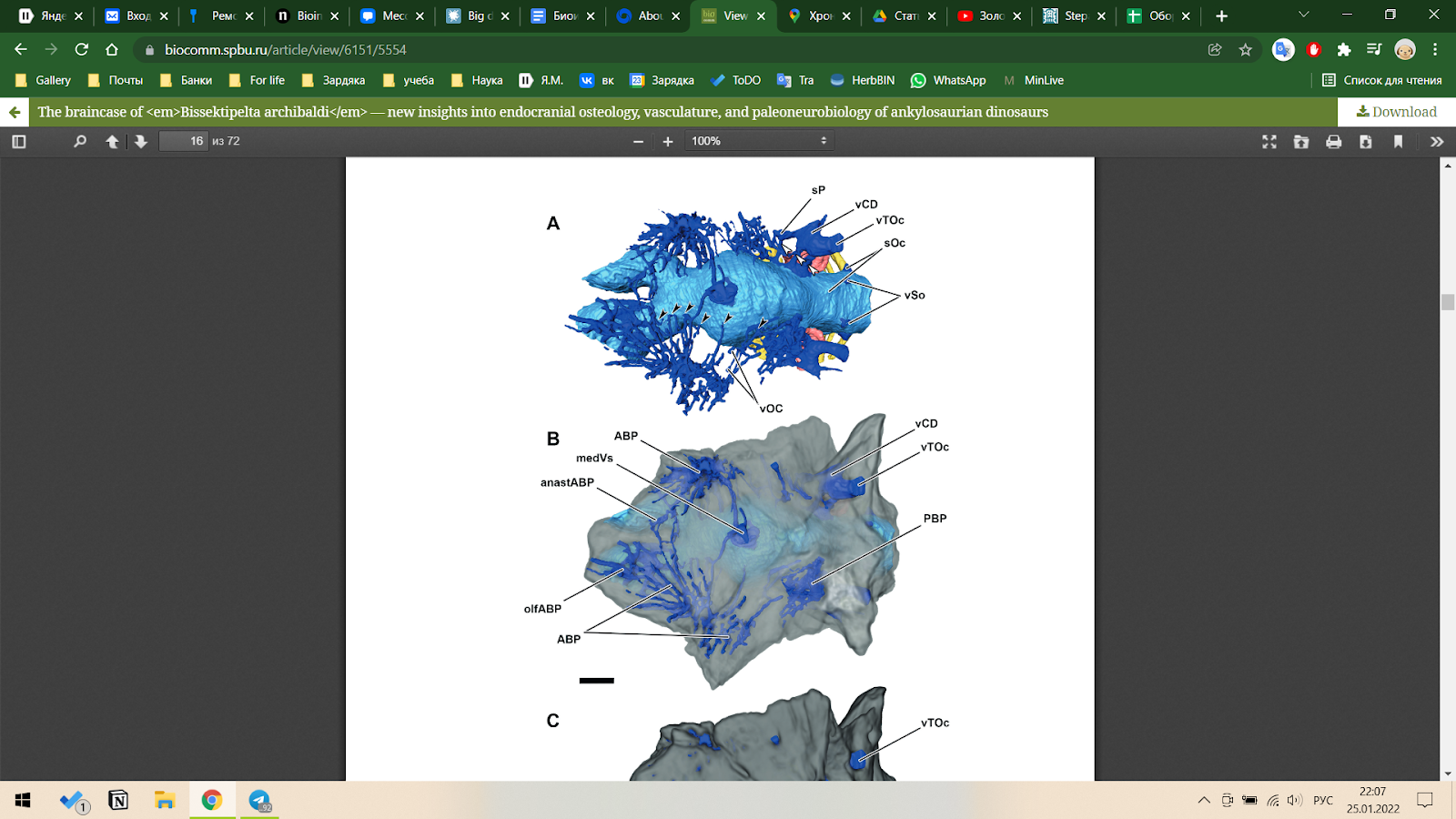
Помимо обработки молекулярных данных, информационные технологии находят своё применение и в традиционных исследованиях. Например, в исследовании по черепной коробке Bissektipelta archibaldi были использованы матрицы признаков, обработка которых полностью переложена на компьютерные вычисления (Kuzmin I. et al., 2020). Также в работе представлены методы 3D моделирования, которые, безусловно, в данном случае представляют собой аналогичные вычислительные методы, без которых порой не обойтись.
Иным примером использования биоинформатики в традиционных исследованиях может послужить орнитологическая работа исследователей из СПбГУ (Bojarinova J. & Babushkina O., 2015). Ещё в 2006 году были начаты эксперименты по влиянию фотопериоду на двигательную активность воробьиных птиц ( сначала длиннохвостых синиц, а после, и иных видов). Отловленные на Ладожской Орнитологической станции птицы помещались в цилиндрические клетки, с установленными камерами и лампами на таймере. Все движения птиц записывались, а отдельная программа обсчитывала, в какую сторону движения птиц были наиболее активными. Безусловно, сбор такого материала мог быть проведён и самими исследователями, путём ежедневного наблюдения за клетками, но это отняло бы гораздо больше времени, а также сильно увеличилась бы погрешность в данных.
Ещё более интересные и сложные примеры можно посмотреть в данной статье. Это сложно, но интересующимся в теме может быть очень занятно..Эти примеры я вставил не просто так. Ведь та часть биоинформатики, которая является разделом биологии, вносит огромный вклад в развитие этой науки. Отдельно стоит отметить роль статистических методов, которые позволяют делать совершенно новые выводы из накопленных данных. Фундаментальные исследования, даже основанные на экспериментальных данных, все больше прибегают к накоплению данных и применению статистических методов обработки этих данных.
Но биоинформатика не панацея. В этой области науки до сих пор остаются нерешённые и актуальные проблемы. Отвлекаясь от частностей, можно сказать, что одной из проблем остаётся верификация прогнозируемых компьютером данных. Так те же предсказания третичной структуры белка (упомянутая выше работа DeepMind) по аминокислотной последовательности получаются лишь при обучении нейросетей на имеющихся данных по весьма коротким аминокислотным последовательностям. В то время как в живых клетках представлены и намного более сложные белки, чьи функции и конформация до сих пор существуют лишь в виде неподтвержденных моделей. И на нынешний момент подтвердить или опровергнуть те или иные модели современными методами невозможно.По итогу, самым насущным вопросом биоинформатики остаётся совершенствование алгоритмов вычислений и разного рода расчётов по моделям, увеличения статистической поддержки тех или иных вычислений и, как ни странно, накопление эмпирических данных традиционными методами биологии. Это всё делается для того, чтобы впоследствии эти данные могли быть применены для обучения разного рода нейросетей.

Нейросеть обучается. Щепотка плоского юмора.
Информационные технологии находят своё применение в построении экологических моделей. И одной из основополагающих работ в этой области можно назвать работу Лотки-Вольтерры. В работе рассматривается закрытый ареал, в котором обитают два вида — травоядные жертвы и хищники. Модель взаимодействия этих видов описывается математическими уравнениями. Конечно, модель эта неточна и имеет много допущений (пищи для травоядных «жертв» в избытке, паразиты и иные хищники отсутствуют, а ареал закрытый. Комплекс этих допущений в живой природе невозможен), но даже такая имитация представляет существенный интерес для биологии как науки и закладывает основы для развития алгоритмов прогнозирования динамики и эволюции разного рода экосистем.
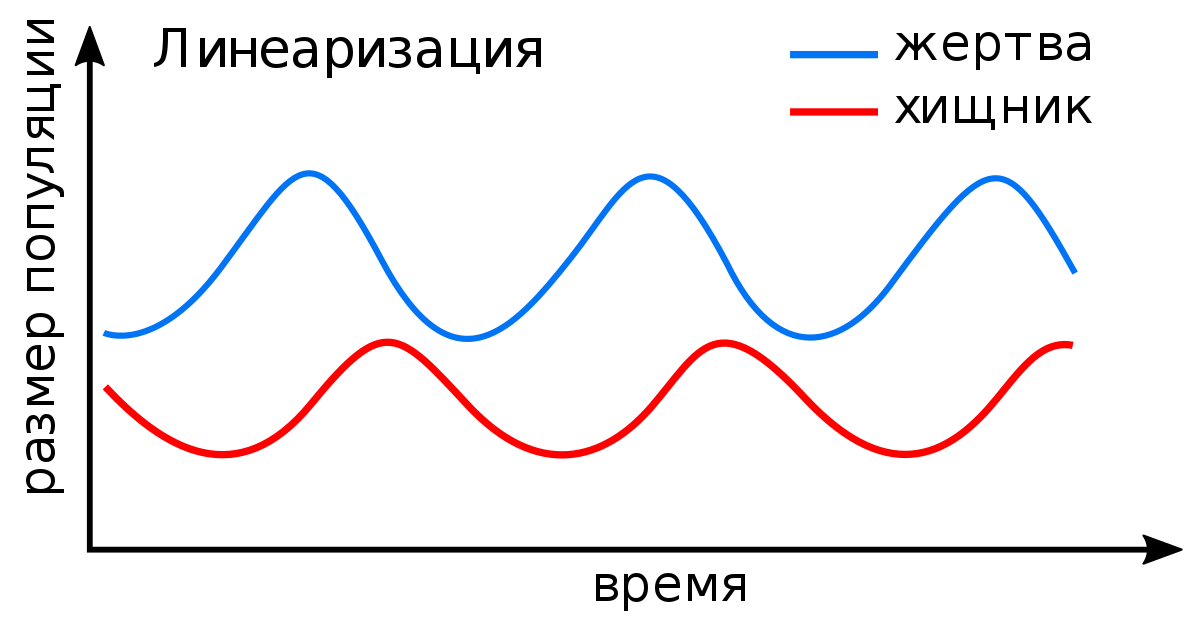 Простая иллюстрация модели Лотки-Вольтерра.
Простая иллюстрация модели Лотки-Вольтерра.Ранее мы уже упоминали, что одно из применений биоинформатика находит в области молекулярной биологии. Работы, основанные на сборке геномов, сравнении и аннотировании и анализе последовательностей, так или иначе, прибегают к методам биоинформатики. Однако же в приближении, оказывается, что решение многих задач берёт своё начало в поиске наибольших совпадений между двумя последовательностями белковыми или нуклеотидными. В случае белковых рассматриваются последовательности аминокислот, в случае нуклеотидных — последовательности нуклеотидов соответственно. Одним из самых первых алгоритмов можно назвать алгоритм Нидлмана–Вунша, разработанный и опубликованный в 1970 году. Иные его названия: алгоритм оптимального соответствия или метод глобального выравнивания. Он применим для выравнивания любых двух строк, будь то аминокислотная, нуклеотидная последовательность или же просто набор букв, цифр.
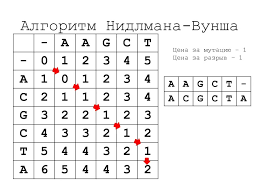
По своей сути компьютер (или человек) даёт оценку всем возможным выравниваниям (соотнося друг с другом последовательности всеми возможными вариантами). И по этой оценки в «баллах» (начисляемых или убавляемых за те или иные варианты) предлагает оптимальный вариант соотнесения представленных строк. Изначально задача может показаться простой, но в случае ДНК(РНК) и белков, вероятны разного рода замены, повторы, инделы (пропуски) в последовательностях, которые могут быть не отражены на сиквенсах (в силу разных причин) и создают большое количество самых разных вариантов соотнесения двух строк. И даже в случае использования мощных компьютеров — такие выравнивания могут занимать не один день.
Несмотря на высокое качество выравниваний при использовании алгоритмов, основанных на алгоритме Нидлмана-Вунша, потраченное на вычисление время, зачастую не соразмерно получаемым данным. И в этом случае на помощь приходят эвристические и вероятностные способы обработки данных. Такие методы не дают высокой точности по сравнению с описанным выше, однако сильно экономят на вычислительной мощности.Эвристические алгоритмы основаны на функции, которая ранжирует альтернативы на каждом шаге ветвления вариантов (ранжирование происходит на основе изначально установленных и введённых данных, то есть, основываясь на уже имеющейся информации) и, таким образом, конечный ответ будет приблизительно соответствовать оптимальному варианту. Такое решение не является абсолютно верным, но остаётся ценным в силу скорости получения ответа. На сходном принципе основана вся эвристика.
Вероятностные же методы, помимо использования основных алгоритмов выравнивания учитывают разные вероятности тех или иных замен. В этом случае строятся так называемые матрицы замен. Разные аминокислоты заменяются в процессе эволюции с разной вероятностью. И для учёта этой неравной вероятности замен используются матрицы. Не вдаваясь в частности, отметим, что и у этих методов есть свои недостатки. Так, точность вероятностных методов сильно уменьшается с увеличением дистанции между таксонами.
WinClada — одна из программ, используемая при работе с матрицами
Мы уже писали про использование матриц признаков выше. Как правило, такой подход используется в морфологических исследованиях. Метод используется в описательной биологии, когда, к примеру, мы хотим установить весь комплекс признаков для 2 или более видов. Безусловно, в приближении идея выглядит довольно просто. Давая количественную или качественную оценку разным признакам у представителей разных видов, мы можем достаточно быстро составить нужную матрицу. Однако же при работе с большими коллекциями или множеством видов (или признаков, а может быть и того и другого) проанализировать получившиеся данные становится крайне затруднительно человеческими силами. И на помощь приходят разные программы для обработки таких данных: TNT, WinClada, PAUP. С помощью них любой человек может провести кладистический анализ имеющихся матриц признаков.
Учитывая разнообразие задач, встающих перед биоинформатиками в разных областях биологии — появление огромного спектра самых разнообразных программ для обеспечения расчётов и анализа моделей — становится неизбежным. Зачастую эти программы не адаптированы для использования обывателем, содержат ошибки, а их оптимизация недостаточно хороша. Примером может служить программа PAST, используемая изначально для обсчёта палеонтологических данных, а в дальнейшем и для решения самых разных статистических задач, притом что там остаются ошибки в алгоритмах некоторых вычислений.Конечно, для стандартных задач уже существует немало понятных программ, с доступным руководством. Большое количество библиотек на таких языках программирования как Python или R. Но всё же круг задач в биологии, для которых использование методов биоинформатике могло бы сильно продвинуть исследования в этих областях — остаётся весьма широк. И востребованность специалистов в этой сфере всё ещё очень высока.
▍Места, где можно обучиться в России. Программы и курсы.
Отдельным плюсом биоинформатики можно отметить её доступность. Практически любой программист или математик может в весьма короткие сроки перепрофилироваться в эту область. Кроме того, любой из наших читателей может познакомиться с некоторыми принципами и методами этой науки. А при большом интересе и найти первую работу в этой области, так как спрос во многом превышает предложение и востребованность таких специалистов велика.
Многие курсы по биоинформатике доступны на разных онлайн-площадках подобных площадке Stepic. Мы предложим лишь несколько курсов, которые нам самим нравятся:
- Этот курс позволяет получить самые начальные навыки в биоинформатике и возможно подтолкнёт новичка к дальнейшему развитию своих навыков;
- А этот курс подойдёт тем, кто хочет получить продвинутые навыки программирования в подсистемах семейства Linux;
- Институт биоинформатики. У них Вы можете найти расширенный список курсов. И, возможно даже поступить на обучение.
▍Заключение.
На множественных примерах мы видим, что биоинформатика является важным разделом в современной биологии. Она оказывает огромное влияние на медицину и биологию, как фундаментальную, так и практическую. Статистические методы открывают небывалые перспективы для многих областей этих наук. При этом новое применение находят многие мониторинговые исследования, не имевшие большого применения в прошлом. Теперь эти огромные массивы данных могут быть проанализированы с применением вычислительной техники. Как это нередко бывает, применение методов на стыке дисциплин и междисциплинарные исследования приносят порой самые неожиданные результаты, которые оказывают большое влияние на развитие науки.
Следует отметить, что биоинформатика до сих принимается некоторыми учёными лишь за метод (к тому же не самый точный) помогающий в исследованиях. И, конечно, у них есть право так считать. На то есть комплекс причин, одна из самых очевидных: несовершенность моделей. Так, на данный момент времени создать идеальную модель той или иной живой системы и учесть все факторы и взаимодействие их влияний — практически невозможно.Мы же, в свою очередь, уверены, что информационные технологии хоть и не являются панацеей, но переводят многие исследования на совершенно новый уровень по скорости и точности исполнения. Это открывает (уже открыло) для человечества совершенно новые перспективы для развития как медицины, так и биологии в целом. А раз так, то мы и не вправе называть биоинформатику просто методом!
Список источников:
- Kuzmin, I., Petrov, I., Averianov, A., Boitsova, E., Skutschas, P., & Sues, H.-D. (2020). The braincase of Bissektipelta archibaldi — new insights into endocranial osteology, vasculature, and paleoneurobiology of ankylosaurian dinosaurs. Biological Communications, 65(2), 85–156. doi.org/10.21638/spbu03.2020.201
- Julia Bojarinova, Olga Babushkina, Photoperiodic conditions affect the level of locomotory activity during autumn migration in the Long-tailed Tit (Aegithalos c. caudatus), The Auk, Volume 132, Issue 2, 1 April 2015, Pages 370–379, doi.org/10.1642/AUK-14-155.1
- deepmind.com/blog/article/alphafold-a-solution-to-a-50-year-old-grand-challenge-in-biology





