
Прежде чем перейти к статье, хочу вам представить, экономическую онлайн игру Brave Knights, в которой вы можете играть и зарабатывать. Регистируйтесь, играйте и зарабатывайте!

Белок бактерии Staphylococcus aureus
В конце ноября команда Google DeepMind объявила о том, что её система глубокого обучения AlphaFold достигла небывалых уровней точности в решении задачи фолдинга белков – трудной проблемы из области вычислительной биохимии.
В чём состоит эта проблема и почему её так трудно решить?
Белки – это длинные цепочки аминокислот. Ваша ДНК кодирует эти последовательности, а РНК помогает производить белки согласно этой генетической схеме. Белки синтезируются в виде линейных цепочек, но впоследствии сворачиваются в сложные шарообразные структуры (см. картинку в начале статьи).
Часть цепочки может свернуться в плотную спираль, "α-спираль". Другая часть может согнуться туда и обратно, сформировав широкую плоскую фигуру, "β-лист":
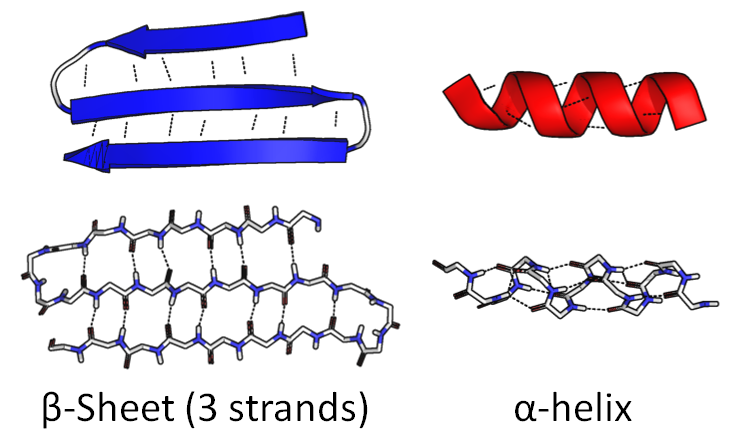
Сама последовательность аминокислот называется первичной структурой. Упомянутые фигуры называют вторичной структурой.
Сами эти компоненты также складываются, формируя уникальные сложные формы. Это называется третичной структурой:
Фермент, взятый у бактерии Colwellia psychrerythraea

Белок RRM3
Выглядит беспорядочно. Почему же этот спутанный клубок аминокислот так важен?
Структура белка не случайна! Каждый белок сворачивается в определённую, уникальную, и по большей части предсказуемую структуру, что совершенно необходимо для его правильной работы. Благодаря физической форме белок хорошо подходит к структурам, с которыми он может связываться. Имеют значение и другие физические свойства, особенно распределение по белку электрического заряда. На картинке положительный заряд обозначен синим, отрицательный – красным:

Поверхностное распределение заряда на белке-переносчике липидов растений 1 риса посевного
Если белок, по сути, представляет собой самособирающуюся наномашину, то основным предназначением последовательности аминокислот будет производство его уникальной формы, распределение заряда, и всё прочее, что определяет функцию белка. Как именно происходит этот процесс, пока не совсем ясно – сегодня это активная область исследований.
В любом случае, понимание структуры важно для понимания её работы. Однако последовательность ДНК задаёт только первичную структуру белка. Как нам узнать его вторичную и третичную структуры – то есть, точную форму, которую примет этот клубок?
Эту задачу называют задачей фолдинга белков, и к ней есть два базовых подхода: измерение и предсказание.
Экспериментальные методы могут измерять структуру белка. Однако это не так просто сделать: в оптический микроскоп структур не видно. Долгое время основным методом исследования структур была рентгеновская кристаллография. Кроме неё, использовался ядерный магнитный резонанс, а в последнее время появилась новая технология, криоэлектронная микроскопия.

Дифракционная рентгеновская картина протеазы SARS
Однако эти методы дороги, сложны и времязатратны, а кроме того, работают не со всеми белками. В частности, белки, встроенные в клеточную мембрану – тот же рецептор ангиотензинпревращающий фермент 2 (ACE2), к которому привязывается вирус COVID-19 – складывается в липидном бислое клетки, и его очень сложно кристаллизовать.

Строение клеточной мембраны
Поэтому мы смогли разобрать структуры крохотного процента от секвенированных белков. В универсальной базе данных белков содержится 180 млн последовательностей, а в базе данных трёхмерных структур белков – всего 170 тысяч позиций.
Нам нужен метод получше.
* * *
Вспомним, что вторичная и третичная структуры белков в основном являются функцией первичной структуры, известной нам благодаря секвенированию. Что если, вместо того, чтобы измерять структуру белка, мы могли бы её предсказать?
Это задача предсказания структуры белков. Специалисты по вычислительной биохимии работают над ней уже несколько десятилетий.
Как к ней можно подступиться?
Очевидный способ – симулировать физику процесса напрямую. Моделируем силы для каждого атома, учитывая его местоположение, заряд и химические связи. Считаем ускорения и скорости, и пошагово прокручиваем эволюцию системы. Это называется «молекулярной динамикой».

Суперкомпьютер "Антон" компании D. E. Shaw Research

Суперкомпьютер IBM Blue Gene
Онлайн-головоломка Foldit
Проблема в том, что такой подход требует чрезвычайно много вычислительных ресурсов. В типичном белке содержатся сотни аминокислот, то есть тысячи атомов. Имеет значение и окружающая среда: при сворачивании белок взаимодействует с окружающей его водой. Поэтому приходится симулировать поведение порядка 30 тысяч атомов. При этом между каждой парой атомов происходит электростатическое взаимодействие, то есть, при грубой оценке, мы получаем 450 млн пар, задачу со сложностью O(N2). Существуют умные алгоритмы, понижающие её сложность до O(N log N). Кроме того, для симуляции необходимо просчитать 109-1012 шагов. Исключительная головная боль.
Хорошо, но нам же не нужно симулировать весь процесс сворачивания. Другой поход предлагает найти структуру с минимальной потенциальной энергией. Обычно объекты склонны приходить в состояние покоя с наименьшей энергией, поэтому такой эвристический подход оправдан. Энергию может подсчитать та же модель молекулярной динамики, что даёт нам величины взаимодействий. С таким подходом мы можем испробовать кучу кандидатов, и выбрать структуру с наименьшей энергией. Проблема, конечно, заключается в том, откуда брать структуры. Их просто слишком много – специалист по молекулярной биологии Сайрус Левинтол подсчитал, что их может быть порядка 10300. Естественно, можно использовать более умный подход, чем случайный перебор. Но их всё равно остаётся слишком много.
Поэтому было предпринято уже множество попыток ускорить подобные вычисления. «Антон», суперкомпьютер от D. E. Shaw Research, использует особое оборудование – специальные интегральные схемы. IBM тоже использует био суперкомпьютер Blue Gene. В Стэнфорде запустили проект Folding@Home, использующий распределённые мощности домашних компьютеров. Проект Foldit от UW превратил фолдинг в игру, чтобы дополнить вычисления интуицией человека.
И всё же долгое время ни одна технология не справлялась с предсказанием широкого спектра белковых структур с большой точностью. На проходящих два раза в год соревнованиях CASP, где результаты работы алгоритмов сравниваются со структурами, измеренными экспериментально, первые места получали предсказания с точностью в 30-40%. До недавнего времени:

Медианная точность предсказаний в категории свободного моделирования у лучшей из команд
Как же работает AlphaFold? Она использует несколько глубоких нейросетей, чтобы обучаться разным функциям, связанным с каждым из белков. Одна из ключевых функций – предсказание итоговых расстояний между парами аминокислот. Это приводит алгоритм к итоговой структуре. В одном из вариантов алгоритма (описанном в журналах Nature и Proteins) была выведена потенциальная функция этого предсказания, к которой был применён простейший градиентный спуск, сработавший на удивление хорошо.
Главное преимущество AlphaFold над предыдущими методами – ему не нужно строить предположения касательно структур. Некоторые методы работают, разбивая белки на участки, просчитывая каждый из них, а потом собирая всё обратно. AlphaFold это не нужно.
Судя по всему, в DeepMind считают проблему фолдинга решённой, что мне кажется излишним упрощением, однако в любом случае их прогресс значителен. Эксперты, не связанные с Google, используют такие эпитеты, как "фантастический" и "революционный".
Теперь у генной инженерии есть уже два мощных инструмента, CRISPR и фолдинг белков. Возможно, 2020-е годы станут для биотехнологий такими же, какими 1970-е были для вычислительной техники.
Поздравляем исследователей из DeepMind с этим прорывом!




